- はじめに (last modified 2018/07/18)
- 参考資料 | 書籍、学会誌 (last modified 2020/05/20)
- 参考資料 | 講習会 (last modified 2020/02/23)
- 参考資料 | 講義、講演資料 (last modified 2020/05/28)
- 過去のお知らせ (last modified 2024/12/02)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2017 (last modified 2017/08/08)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2016 (last modified 2016/08/22)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2015 (last modified 2016/08/19)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGS速習コース2014 (last modified 2015/02/11)
- 書籍 | トランスクリプトーム解析 | について (last modified 2014/05/12)
- 書籍 | トランスクリプトーム解析 | 1.1 はじめに (last modified 2014/05/09)
- 書籍 | トランスクリプトーム解析 | 1.2.1 原理(Affymetrix 3'発現アレイ) (last modified 2014/05/12)
- 書籍 | トランスクリプトーム解析 | 1.2.2 最近の知見 (last modified 2014/05/09)
- 書籍 | トランスクリプトーム解析 | 2.2.1 生データ(プローブレベルデータ)取得 (last modified 2014/04/18)
- 書籍 | トランスクリプトーム解析 | 2.2.2 データの正規化(基礎) (last modified 2014/04/17)
- 書籍 | トランスクリプトーム解析 | 2.2.3 データの正規化(計算例) (last modified 2014/04/18)
- 書籍 | トランスクリプトーム解析 | 2.2.4 データの正規化(その他) (last modified 2014/04/18)
- 書籍 | トランスクリプトーム解析 | 2.2.5 アノテーション情報 (last modified 2014/04/18)
- 書籍 | トランスクリプトーム解析 | 2.3.1 RNA-seqデータ(FASTQファイル) (last modified 2016/03/17)
- 書籍 | トランスクリプトーム解析 | 2.3.2 リファレンス配列 (last modified 2014/04/16)
- 書籍 | トランスクリプトーム解析 | 2.3.3 アノテーション情報 (last modified 2017/08/02)
- 書籍 | トランスクリプトーム解析 | 2.3.4 マッピング(準備) (last modified 2014/06/20)
- 書籍 | トランスクリプトーム解析 | 2.3.5 マッピング(本番) (last modified 2014/06/21)
- 書籍 | トランスクリプトーム解析 | 2.3.6 カウントデータ取得 (last modified 2016/02/09)
- 書籍 | トランスクリプトーム解析 | 3.2.1 クラスタリング(データ変換や距離の定義など) (last modified 2014/05/16)
- 書籍 | トランスクリプトーム解析 | 3.2.2 実験デザイン, データ分布, 統計解析との関係 (last modified 2014/04/18)
- 書籍 | トランスクリプトーム解析 | 3.2.3 多重比較問題 (last modified 2014/04/19)
- 書籍 | トランスクリプトーム解析 | 3.2.4 各種プロット(M-A plotや平均-分散プロットなど) (last modified 2014/04/19)
- 書籍 | トランスクリプトーム解析 | 3.3.1 解析目的別留意点 (last modified 2014/04/20)
- 書籍 | トランスクリプトーム解析 | 3.3.2 データの正規化(基礎編) (last modified 2014/06/23)
- 書籍 | トランスクリプトーム解析 | 3.3.3 クラスタリング (last modified 2014/04/20)
- 書籍 | トランスクリプトーム解析 | 3.3.4 各種プロット (last modified 2014/04/27)
- 書籍 | トランスクリプトーム解析 | 4.2.1 2群間比較 (last modified 2014/04/19)
- 書籍 | トランスクリプトーム解析 | 4.2.2 他の実験デザイン(paired, multi-factor, 3群間) (last modified 2014/04/19)
- 書籍 | トランスクリプトーム解析 | 4.2.3 多群間比較(特異的発現パターン) (last modified 2014/04/20)
- 書籍 | トランスクリプトーム解析 | 4.3.1 シミュレーションデータ(負の二項分布) (last modified 2014/04/27)
- 書籍 | トランスクリプトーム解析 | 4.3.2 データの正規化(応用編) (last modified 2014/04/27)
- 書籍 | トランスクリプトーム解析 | 4.3.3 2群間比較 (last modified 2014/04/28)
- 書籍 | トランスクリプトーム解析 | 4.3.4 他の実験デザイン(3群間) (last modified 2014/04/28)
- 書籍 | 日本乳酸菌学会誌 | について (last modified 2020/05/20)
- 書籍 | 日本乳酸菌学会誌 | 第1回イントロダクション (last modified 2018/09/03)
- 書籍 | 日本乳酸菌学会誌 | 第2回GUI環境からコマンドライン環境へ (last modified 2018/09/03)
- 書籍 | 日本乳酸菌学会誌 | 第3回Linux環境構築からNGSデータ取得まで (last modified 2017/07/02)
- 書籍 | 日本乳酸菌学会誌 | 第4回クオリティコントロールとプログラムのインストール (last modified 2018/05/10)
- 書籍 | 日本乳酸菌学会誌 | 第5回アセンブル、マッピング、そしてQC (last modified 2017/06/25)
- 書籍 | 日本乳酸菌学会誌 | 第6回ゲノムアセンブリ (last modified 2017/06/21)
- 書籍 | 日本乳酸菌学会誌 | 第7回ロングリードアセンブリ (last modified 2017/06/28)
- 書籍 | 日本乳酸菌学会誌 | 第8回アセンブリ後の解析 (last modified 2017/06/28)
- 書籍 | 日本乳酸菌学会誌 | 第9回ゲノムアノテーションとその可視化、DDBJへの登録 (last modified 2017/03/13)
- 書籍 | 日本乳酸菌学会誌 | 第10回DDBJへの塩基配列の登録(後編) (last modified 2017/06/28)
- 書籍 | 日本乳酸菌学会誌 | 第11回統合データ解析環境Galaxy (last modified 2017/11/13)
- 書籍 | 日本乳酸菌学会誌 | 第12回Galaxy:ヒストリーとワークフロー (last modified 2018/07/04)
- 書籍 | 日本乳酸菌学会誌 | 第13回RNA-seq解析(その1) (last modified 2022/05/01)
- 書籍 | 日本乳酸菌学会誌 | 第14回RNA-seq解析(その2) (last modified 2022/05/01)
- 書籍 | 日本乳酸菌学会誌 | 第15回RNA-seq解析(その3) (last modified 2022/12/18)
- 書籍 | 日本乳酸菌学会誌 | 第16回なぜ次から次へと新規手法が開発されるのか? (last modified 2021/08/16)
- 書籍 | 日本乳酸菌学会誌 | 第17回バイオインフォマティクス教育の今後 (last modified 2022/05/01)
- 書籍 | 日本乳酸菌学会誌 | 第18回遺伝子発現データのクラスタリング (last modified 2022/12/18)
- 書籍 | 日本乳酸菌学会誌 | 第19回R Markdown (last modified 2022/11/30)
- 書籍 | 日本乳酸菌学会誌 | 第20回RNA-seqカウントデータの性質と統計モデル (last modified 2022/12/19)
- 書籍 | 日本乳酸菌学会誌 | 第21回シミュレーションデータの生成 (last modified 2025/06/21)
- 書籍 | 日本乳酸菌学会誌 | 第22回メタゲノム解析(その1) (last modified 2024/02/09)
- 書籍 | 日本乳酸菌学会誌 | 第23回メタゲノム解析(その2) (last modified 2024/03/04)
- 書籍 | 日本乳酸菌学会誌 | 第24回メタゲノム解析(その3) (last modified 2024/09/10)
- 書籍 | 日本乳酸菌学会誌 | 第25回メタゲノム解析(その4) (last modified 2024/12/02)
- 書籍 | 日本乳酸菌学会誌 | 第26回サンキー図・沖積図・パラレルセット (last modified 2025/03/31)
過去のお知らせ
- 2024年
- 「書籍 | 日本乳酸菌学会誌 | 第22回」と「書籍 | 日本乳酸菌学会誌 | 第23回」のリンク先を修正しました。(2024/06/30)
- 2023年
- アグリバイオインフォマティクスの教科書「Web連携テキスト バイオインフォマティクス」のページの読み込みに時間がかかる問題がありました。理由は1つの巨大なページとして構成していたためですが、章ごとのページに変更することで解決しました。(2023/05/24)
- 「書籍 | 日本乳酸菌学会誌 | 第20回」の原稿PDFを公開しました。(2023/04/01)
- 2023年4月4日18:00-のアグリコクーン全体ガイダンスの冒頭部分で、アグリバイオインフォマティクスの簡単な紹介をさせていただきます。(2023/03/24)
- 「書籍 | 日本乳酸菌学会誌 | 第19回」の原稿PDFをを公開しました。(2023/01/13)
- 2022年
- 「(Rで)マイクロアレイデータ解析」のところに取り残されていた2014年出版のトランスクリプトーム解析という本のWeb資料情報をこちらに移行しました。古いのでもう見ることもないと思っていましたが、p121-129の3.2.4項やp145-165の3.3.4項の平均-分散プロットなどを最近見返していたなかで、今回のミスに気づいた次第です。(2022/12/24)
- 「書籍 | 日本乳酸菌学会誌 | 第15回RNA-seq解析(その3)」のCPM, CPK, FPKM, and TPMのところで余分なhtmlタグがあったのを修正しました。(2022/12/18)
- バイオDBとウェブツール ラボで使える最新70選(小野浩雅 編)が出版されています。私がよくお世話になっているTogo picture galleryなどいろいろありますが、ざっくりと最新状況を俯瞰できてトータルで有用という位置づけだと思っています。今後も3年ごとくらいに定期的に出版されるとありがたいです。(2022/11/06)
- アグリバイオインフォマティクスの教科書「Web連携テキスト バイオインフォマティクス」が培風館より刊行されました。タイトルのWeb連携に相当する部分はこちらです。(2022/10/26)
- 日本乳酸菌学会誌の第18回の原稿を公開しました。(2022/08/25)
- 「実験医学別冊 論文図表を読む作法」が出版されています。タイトル通りですが、私個人としてはAccumulation curveの解説を入れていただいて大変助かっております。これまでなかなかとっつきにくかった図の理解が進む良書だと思います。(2022/07/27)
- 日本乳酸菌学会誌の第16回と第17回の原稿をこちらでも公開しました。(2022/05/29)
- 「書籍 | 日本乳酸菌学会誌 | 第17回バイオインフォマティクス教育の今後 」を追加しました。第16回と17回ともに原稿はまだ公開されていませんが、そのうち公開されると思います。(2022/05/01)
- 「書籍 | 日本乳酸菌学会誌 | ...」の第13回から15回までのウェブ資料PDFがサーバ移行のどさくさでリンク切れになっていたので修正しました。(2022/05/01)
- 東京大学・大学院農学生命科学研究科・応用生命工学専攻の令和5(2023)年度大学院学生募集公開ガイダンスは、2022年5月7日(土)と5月28日(土)に開催します。(2022/04/17)
- 「書籍 | 日本乳酸菌学会誌 | 第15回RNA-seq解析(その3)」のW11から13にかけての提供ファイル(例:Kallisto_9samples.xlsx)のリンク切れを修正しました。(2021/07/22)
- 2021年
- 独習 Pythonバイオ情報解析が2021年3月に出版されています。 一般的なプログラミング言語として解説から、塩基配列データの取り扱い、データの可視化、そしてRNA-seq解析周辺など、 非常に豊富な内容となっています。編集代表の黒川顕先生にはNGSハンズオン講習会の最終年度でお世話になり、執筆者の多くの先生にはアグリバイオインフォマティクス教育研究プログラム関連講義でもお世話になっております。(2021/03/27)
- Dr.Bonoの生命科学データ解析 第2版が2021年3月に出版されています (バイオインフォマティクス初学者向けの本)。前回の第1版から3年以上経過しており、WindowsでのLinux環境(WSL2)の話など最新情報に アップデートされているのが基本形です。しかし、大枠として変わってない部分もさらっとでも読むとよいと思います。 第1版当時の自分には無縁で記憶に残っていない事柄でも、今の自分と関係があるかもしれないからです (私の場合はそれがオーソログクラスターでした)。(2021/03/18)
- 令和3年度のアグリバイオインフォマティクス教育研究プログラム に関する情報をトップページに掲載しています。(2021/03/18)
- 2020年
- R2年度はもうキャパオーバーですので、ご新規様の講演や執筆依頼はお控えくださいますようお願い申し上げます。(2020/09/19)
- 2020年11月14-15日(土日)に数理生物学セミナー2020@TMDUというオンラインセミナーが開催されます。 興味ある方はどうぞ。(2020/09/19)
- single-cell RNA-seq (scRNA-seq)の解析パイプラインのガイドラインに関する論文であるVieth et al., Nat Commun., 2019についての批評論文が公開されました (Kadota and Shimizu, Front Genet., 2020)。特にscRNA-seqをbulk RNA-seqと差別化する際の論法や、 比較対象として用いたbulk RNA-seq用の正規化法の選定に関して、論文調査不足・事実誤認・ミスリード・不誠実さといった観点で痛烈に批判しています。(2020/07/28)
- 2019年
- 「生命科学者のためのDr.Bonoデータ解析実践道場(著:坊農秀雅)」が出版されています。 今回の"Bono本"は、アグリバイオの大学院講義で丁寧に教えることが現実的に難しいLinux環境でのデータ解析の情報が丁寧に解説されています(アグリバイオの内容と相補的な関係)。 「聞いたことはあるがよく知らない事柄」が簡潔かつ丁寧に書かれているので、私は主にそのあたりの頭の整理に利用させてもらっています。(2019/09/30)
- 「参考資料 | 講義、講演資料」中のリンク切れを修正しました。(2019/08/02)
- 「進化で読み解く バイオインフォマティクス入門(著:長田直樹)」が出版されています。 本書の何よりも素晴らしいところは、単著だという点だと思います(統一感って重要)。そしてチャラチャラしたところがなく、中身がしっかりしており、 そして幅広い内容が丁寧に解説されているという点が非常によいと思います。(2019/07/05)
- 「参考資料 | 講義、講演資料」を更新しました。(2019/04/10)
- 乳酸菌学会誌のNGS連載第13回のウェブ資料PDFのW2のFASTA形式ファイルで保存する部分、よりよい方法が見つかったのでそれに変更しました。(2019/04/05)
- 日本乳酸菌学会誌のNGS関連連載の第13回分原稿PDFを公開しました。ウェブ資料も公開しました。(2019/03/18)
- RNA-seqカウントデータ解析用RパッケージであるTCCのGUI版である、 TCC-GUIの論文がpublishされました。(2019/03/14)
- TCC-GUIのオンライン版の基本的な利用法は、2019年3月15日予定の講義資料(の後半部分)に記載しています。 このオンライン版はインストールが不要という長所がある一方で、すぐに接続が切れる短所があります。これを回避するローカル版の起動方法(RStudioのインストール含む)も講義資料(の最後のほう)に記載しています。(2019/03/03)
- 「参考資料 | 講義、講演資料」を更新しました。(2019/03/02)
- 2018年
- 2018年11月に「よくわかるバイオインフォマティクス入門(藤博幸 編)」という本が出ました。(2018/11/21)
- 2019年度もアグリバイオインフォマティクス教育研究プログラムを実施します。 例年東大以外の企業の方、研究員、大学院生が2割程度受講しております。受講ガイダンスは、2019年4月5日(Fri.)17:15より東大農学部2号館2階化学第一講義室で開催します。(2019/03/11)
- TCCのオンラインGUI版(TCC-GUI)を公開しました。 volcano plotなど図の作成までやってくれる上に、それがどのようなスクリプトで書かれているかも理解できる仕様になっています。 バグレポートやリクエストなどをいただければ幸いですm(_ _)m(2018/10/15)
- 平成29年度のNGSハンズオン講習会でもお世話になった「先進ゲノム支援」による中級者向けの 情報解析講習会が2018年11月19-21日に開催されます。(2018/09/20)
- 「書籍 | 日本乳酸菌学会誌 | 第2回GUI環境からコマンドライン環境へ」の最後、 および「書籍 | 日本乳酸菌学会誌 | 第3回Linux環境構築からNGSデータ取得まで」の最初のほうにある、 「1. VirtualBox、および2. Extension Packのインストール手順」のWindows版を更新しました。(2018/09/03)
- 参考資料 | 講義、講演資料を更新しました。(2018/07/21)
- 日本乳酸菌学会誌のNGS関連連載の第12回分原稿PDFを公開しました。ウェブ資料も公開しました。(2018/07/04)
- H29年度NGSハンズオン講習会の報告書が公開されました。(2018/05/17)
- 平成29年度NGSハンズオン講習会の動画が公開されています。(2018/03/01)
- 2017年
- 日本乳酸菌学会誌のNGS関連連載の第11回分原稿PDFを公開しました。ウェブ資料も公開しました。(2017/11/13)
- 平成29年度NGSハンズオン講習会の森先生講義資料PDF。(2017/08/30)
- 平成29年度NGSハンズオン講習会の8/31および9/1出席者は、インポートしたovaを起動して、 キーボード入力確認PDF(約2MB)」に従って、正しく日本語キーボードでの入力が可能かどうかを確認しておいてください。(2017/08/28)
- 平成29年度NGSハンズオン講習会の門田担当分の「プラスアルファの内容からなる補足資料PDF(約3MB)」を追加しました。(2017/08/08)
- 平成29年度NGSハンズオン講習会の門田担当分のスライドPDF(約14MB)を2017.08.04版にマイナーアップデートしました。(2017/08/04)
- 「書籍 | トランスクリプトーム解析 | 2.3.3 アノテーション情報」の一番最初のp78の網掛け部分において、 フィルタリング条件の指定のところを"with_ox_refseq_mrna"から"with_refseq_mrna"に変更しました (小松将大氏 提供情報)。3年も経過しているので、おそらく他にも沢山使えなくなっているオプションや関数があるのでしょうね。可能な範囲で情報提供お願いしますm(_ _)m(2017/08/02)
- 平成29年度NGSハンズオン講習会の門田担当分コピペ用コード集がほぼ完成しています。 このような流れで話す予定です。(2017/06/28)
- 日本乳酸菌学会誌のNGS関連連載の第10回分原稿PDFを公開しました。(2017/06/28)
- 平成28年度NGSハンズオン講習会の 報告書 (約3MB)が公開されました。(2017/04/20)
- 平成29年度NGSハンズオン講習会の ウェブページが公開されました。受講申込の受付期間は2017年5月15日(月)14時00分~6月23日(金)12時00分です。(2017/05/15)
- 日本乳酸菌学会誌のNGS関連連載の第9回分原稿PDFを公開しました。(2017/03/13)
- 「書籍 | 日本乳酸菌学会誌 | 第7回ロングリードアセンブリ」でgrep -Aの説明が間違っていたので修正しました。 「検索文字列の行を含む後ろx行を表示」みたいに書いていましたが、検索文字列の行は含まないようですm(_ _)m(2017/01/26)
- NGSハンズオン講習会2016の2016年7月22日の講義資料のスライド107で、 effective library sizesからのサイズファクター(size factors)への変換式がef.libsizes/mean(ef.libsizes)となっています。 じつはこれって間違いで本当はmean(ef.libsizes)/ef.libsizesだったんじゃないかと、、、ふと思い始めています。 当時誰からも指摘されませんでしたが(話の流れ的にはTips的なところだったからかもしれません)、 ヘビーユーザの方は特にTCC正規化係数を他のパッケージ(特にsize factorsに変換して使う場合は)と組み合わせる場合は、ちょっと気にかけておいてください。 間違いが確定であると思われた方はレポートいただければ幸いですm(_ _)m。(2017/01/04)
- NGSハンズオン講習会2016の講義映像 (統合TVと Youtube)が公開されました。(2016/12/07)
- 日本乳酸菌学会誌のNGS関連連載の第8回分原稿PDFを公開しました。(2016/11/30)
- バイオインフォマティクスやNGSをキーワード程度は知っているヒト(学部3年生程度)向けの講義資料を作成しました。 「参考資料 | 講習会、講義、講演資料」の2016.09.12-16の日付のものです。 日本乳酸菌学会誌のNGS連載第1-4回あたりのダイジェスト版のようなものです。 NGSハンズオン講習会2016の予習事項が厳しすぎて受講を断念したヒトも、 ここからだとスムーズに頭に入っていくかもしれません。(2016/08/26)
- NGSハンズオン講習会2016の「昨年度との違い、対応関係、想定受講者、および予習事項」の PDFに注釈を入れました。 8/4のcuffdiffの入力ファイル周辺の話です。経緯などの記憶は定かではありませんが、 ここで誤解を与えていたかもしれないと思いましたので、念のため追記しました。 これ以外にも間違いや私の誤解などありましたら、よりよい情報共有のために遠慮なくご指摘よろしくお願いいたしますm(_ _)m。(2016/08/19)
- NGSハンズオン講習会2016の第3部(8/4)の講義資料PDFを修正しました。 スライド107 (Trinityのパスを通すところのミス)、スライド140 (Bridgerに必要なboostパッケージをapt-getでやるとサンプルデータの実行もうまくいく)あたりです。(2016/08/12)
- NGSハンズオン講習会2016の第2部(7/25-28)の講義資料修正版を公開しました。(2016/08/03)
- NGSハンズオン講習会2016の第3部(8/01-04)の講義資料を一部微修正しました。(2016/07/31)
- Twitterハッシュタグ AJACSはこちら。(2016/07/27)
- NGSハンズオン講習会2016の第2部(7/25-28)の コピペ用コマンド集も公開しました。 当日までにアップデートする可能性はあります。(2016/07/21)
- NGSハンズオン講習会2016の第2部および第3部出席予定で貸与PCの方へのお知らせ。 結局講習会期間を通して同じPCを使うことになりましたので、運営側では第1部終了後にovaファイルの再インポートは行わないことになりましたので、 必要に応じて各自で行ってください。(2016/07/21)
- NGSハンズオン講習会2016の第2部(7/25-28)の講義資料を公開しました。(2016/07/20)
- NGSハンズオン講習会2016の第1部(7/20-22)は、各日のウェブサイト( 2016.07.20; 2016.07.21; 2016.07.22) をスクロールしながら進めていきますが、スライド番号との対応づけを行いました。 第1部の心構え(掟)ではやらないと書いていた気がしますが、 やらずにこれに起因する迷える子羊が出たときに後悔しないようにするためです。(2016/07/13)
- NGSハンズオン講習会2016の第1部(7/20-22)の講義資料PDFについて、 全体の微修正や統一化を7/12-13に行いましたのでバージョンが2016.07.12版および2016.07.13版に変わっています。 印刷版は2016.06.23版あたりのままになっておりますがご了承くださいm(_ _)m (2016/07/13)
- 2016年7月25日18:30-20:30に開催されるNGSハンズオン講習会2016の情報交換会(アメリエフ主催)の 申込みフォームができました。 会場は東大農学部キャンパス内のアブルボアです。 締切は7/20の12:00までです。(2016/07/12)
- NGSハンズオン講習会2016受講生の方へ。 PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB) をしっかり見ておいてください。(2016/07/09)
- NGSハンズオン講習会2016の第1部と第3部の講義資料が出揃いました。(2016/07/07)
- 日本乳酸菌学会誌のNGS関連連載の第7回分原稿PDFを公開しました。(2016/07/01)
- 上記メール不具合に関連して、平成28年度ハンズオン講習会受講申込みをされた方で、 まだNBDC事務局から何の連絡も受けていないという方も、その旨をアグリバイオ事務局 and/or 「koji.kadota @ gmail.com」宛てにご連絡ください。(2016/06/15)
- 日本乳酸菌学会誌のNGS関連連載の第7回ウェブ資料PDFを公開しました。(2016/05/12)
- 平成28年度ハンズオン講習会が7/19-8/4の日程で開催されます。 申込み期間は4/4-5/31です。追加申込みやキャンセル待ちの受付は絶対に行いませんので受講希望者はお気をつけください。 門田担当部分は主にこれまでの受講生を対象としています(リクエストを多く反映させています)。 「昨年度との違い、対応関係、想定受講者、および予習事項」については こちらのPDF (約2MB) をご覧になり、必要に応じてつまみ食いしてください。(2016/04/05)
- 日本乳酸菌学会誌のNGS関連連載の第6回分原稿PDFを公開しました。(2016/04/21)
- 日本乳酸菌学会誌のNGS関連連載の 第6回ウェブ資料を更新しました。具体的には、W20-2のスライド256の解説内容を変更しました。(2016/03/29)
- 「書籍 | トランスクリプトーム解析 | 2.3.1 RNA-seqデータ(FASTQファイル)」 のgetFASTQinfoやgetFASTQfile関数実行部分でsra_conという引数を追加しました。 が、おそらくデータを取りに行っているENAのURL(http://www.ebi.ac.uk/ena/data/view/reports/sra/fastq_files/)がリンク切れ になっているようでうまく情報の取得ができなくなっているようですm(_ _)mもちろん私のせいではありませんw。ご指摘深謝m(_ _)m(2016/03/17)
- 2016年3月3-4日に行われるHPCI講習会・バイオインフォマティクス実習コースの受講生 (およびキャンセル待ち受講希望者)の皆様へ。実習用hogeフォルダのzip圧縮ファイルhoge.zip(約150MB; 20160222, 17:52版)をアップしました。 講義資料PDF(約16MB; 2016.03.05版)もhogeフォルダ中にあります。(2016/03/05)
- 平成27年度NGSハンズオン講習会の 報告書が公開されているようです。(2016/02/22)
- 日本乳酸菌学会誌のNGS関連連載の 第6回ウェブ資料をアップしました。(2016/02/03)
- 日本乳酸菌学会誌のNGS関連連載の第5回ウェブ資料を更新しました。 2015年12月下旬に一気に全てやり直したので、若干プログラムのバージョンが上がっています。(2015/12/22)
- 日本乳酸菌学会誌のNGS関連連載の第4回ウェブ資料を更新しました。 2015年12月初旬に一気に全てやり直したので、若干プログラムのバージョンが上がっています。 各回終了時点のovaファイル(約 6 GB)も提供可能です。(権利関係上無条件公開はできませんので...)欲しい方は、メールの タイトルを「乳酸菌連載第x回終了時点のovaファイル希望」として私宛にメールしてください(本文は空でOK)。URLをお知らせします。(2015/12/11)
- (お知らせをすっかり忘れてましたが)HPCI講習会・バイオインフォマティクス実習コースの 2016年3月3-4日分を担当します。Tang et al., BMC Bioinformacis, 2015 の内容を含められればと思っています。興味ある方はどうぞ。(2015/12/07)
- 日本乳酸菌学会誌のNGS関連連載の第3回ウェブ資料を更新しました。 オリジナル版は約1.35億リードからなる計約15GBのpaired-end RNA-seqデータのダウンロードについて記載していましたが、 こけたヒトが一定数いらっしゃったようですので、ダウンロードの段階で最初の150万リードのみしかダウンロードしないやり方に変更しました。(2015/12/07)
- 日本乳酸菌学会誌のNGS関連連載の第2回で示したのは isoファイルからのBio-Linux 8のインストール手順だけでしたが、ovaファイルからのインストール手順(Windows版とMacintosh版両方) も示しました。ここのやり方を参考にしていただければ、例えば(要望に応じて門田が提供する)連載第4回終了時点のPC環境を保存したovaファイルなどを自分でインポートすることで、 エンドユーザは第5回以降の内容を独立して自習することができます。(2015/11/26)
- 日本乳酸菌学会誌のNGS関連連載の第2-3回辺りで、 VirtualBoxとBio-Linux 8のインストール手順を(Windows版とMacintosh版両方)アップデートしました。 情報量は若干増やして、ovaファイルの作成なども追加しました。(2015/11/19)
- 日本乳酸菌学会誌のNGS関連連載の第5回分PDFを公開しました。(2015/11/17)
- NGSハンズオン講習会の講義映像・動画が公開されました。(2015/11/13)
- 日本乳酸菌学会誌のNGS関連連載の第4回分ウェブ資料PDFにおいて、 2015年9月18日に別のノートPCで、共有フォルダ関連の「sudo apt-get install dkms」(W4-5-2)実行時にエラーに遭遇したので、対処法を記載しました。(2015/09/18)
- NGSハンズオン講習会2015のアメリエフ様分(服部先生と山口先生)の講義資料を差し替えました。(2015/09/03)
- NGSハンズオン講習会のフォローアップ勉強会(アメリエフ主催)が2015年9月18日(金)に開催されます。 申込み締切は9月14日(月)正午までです。参加希望者は申込みフォームよりお申込みください。 尚、場所はJR神田駅周辺の居酒屋wですのでお気軽にご参加ください。私も参加予定ですw。(2015/09/02)
- 日本乳酸菌学会誌の連載第5回を書き始めています。公開データDRR024501 (paired-end MiSeqデータ) のゲノムアセンブリ手順を書いていますが、ちょっとイマイチな感じです(配列数約3万)。 MiSeqデータのみで(もちろん合理的な手順で)もう少し配列数を減らしたいと思っていますので、より良いと思われる手順をお知らせいただければ幸いです。 と書いていましたが原著論文(Tanizawa et al., BMC Genomics, 2015)著者から詳細情報をいただき、 k=31でアセンブルしたときは同じような結果になったということです。k=200あたりでよりよい結果になったということです。 MiSeqは250 bp程度の長さなので確かにこれくらい大きめのkにしないといけないですね。連載第5 or 6回で詳細に述べる予定です。(2015/08/27)
- 日本乳酸菌学会誌のNGS関連連載の第4回分ウェブ資料PDF中の、 共有フォルダ関連の記述(W4-5から4-8周辺)をアップデートしました。8/14にお知らせしましたが、8/23に再びMacとWinを統一的に行うやり方に変更しました。 これでもまだ設定がリセットされるという方はお手数ですがお知らせ願います。(2015/08/26)
- 日本乳酸菌学会誌のNGS関連連載の第4回分PDFを公開しました。(2015/08/19)
- 日本乳酸菌学会誌のNGS関連連載の第4回分ウェブ資料PDF中の、 共有フォルダ関連の記述(W4-5から4-8周辺)をアップデートしました。これでもまだ設定がリセットされるという方はお手数ですがお知らせ願います。(2015/08/14)
- 2015年8月26-28日開催のNGSハンズオン講習会2015の 追加受講申込を受け付けることにしました(複数受講希望問合せがあり、2回に分けたので若干ゆとりがあるのでそうしました)。締め切りは8月18日(火)12時00分だそうです。(2015/08/07)
- 2015年7月22日-8月6日開催のNGSハンズオン講習会2015の 講義資料(8/6森岡先生分)を公開しました。「2015.08.06」でページ内検索してください。(2015/08/05)
- 2015年7月22日-8月6日、および8月26-28日開催のNGSハンズオン講習会2015の 講義資料(8/5後半のRNA-seq統計解析部分の最終版)を公開しました。(2015/08/03)
- NGSハンズオン講習会2015の2015年8月3-5日のどこかで利用予定の コマンドファイル(command.zip)をダウンロードしておいてください。 中身はReseq_command.txtと RNA-seq_command.txtです。 場所はどこでもいいですが、ゲストOS上が無難かもしれません。(2015/07/28)
- 2015年7月22日-8月6日、および8月26-28日に開催予定のNGSハンズオン講習会2015の 講義資料(門田の7/23のLinux最終版と7/29のR最終版)を公開しました。(2015/07/22)
- 2015年7月22日-8月6日、および8月26-28日に開催予定のNGSハンズオン講習会2015の 講義資料(アメリエフ様分と門田分の一部)を公開しています。(2015/07/17)
- 2014年9月1-12に開催されたNGS速習コースの報告書PDF(3.7MB)がNBDCのサイトで公開されています。(2015/07/01)
- 平成27年度NGSハンズオン講習会を2015年7月22日-8月6日の11日間で実施します。 受講生の申込期間は5月15日(金)12時00分で終了しました。しかし、 部分受講可能にした副作用としてボランティアの講義補助要員(TA)数が非常に少なくピンチです。 申込み受付サイトでTAのみ7月10日まで募集を延長します。 可能な範囲でご協力をお願いしますm(_ _)m(2015/05/15)
- 日本乳酸菌学会誌のNGS関連連載の第3回分PDFを公開しました。(2015/04/20)
- 平成27年度NGSハンズオン講習会を2015年7月22日-8月6日の11日間で実施します。 受講申込期間は4月20日(月)-5月15日(金)12時00分です。 申込み受付サイトで受講生とTAの募集の両方を行っております。 昨年度の「NGS速習コース」 同様、オブザーバー(TA)募集も行っていますので可能な範囲でご協力をお願いしますm(_ _)m(2015/04/21)
- 2015年03月05-06日に産総研・臨海副都心センターでRのハンズオン講習会(Rでゲノム・トランスクリプトーム解析:CpG解析から機能解析まで)が開催されます。 キャンセル待ち受付は2015年2月3日(火)午前11時を予定しているそうです。(2014/12/05)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGS速習コースの動画が統合TVより公開されました。 速習コース全体を俯瞰できるYoutubeのリストはこちら。(2014/12/04)
- 門田幸二 著シリーズ Useful R 第7巻 トランスクリプトーム解析、およびこのウェブページ中で頻用させていただいているQuasR パッケージの原著論文(Gaidatzis et al., Bioinformatics, 2015)が公開されたので関連個所をアップデートしました。(2014/12/03)
- 日本乳酸菌学会誌のNGS関連連載の一環としてBio-Linux 8のインストール手順(Win用とMac用)を作成しました。 詳細は書籍 | 日本乳酸菌学会誌 | についてをご覧ください。(2014/11/27)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGS速習コース。 9/11の河岡先生の講義資料、用語解説などを追加したバージョンをいただきました。(2014/11/06)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | 速習コース。 9/4の服部先生のperlプログラム(perl10.pl)の修正版、およびWindowsで作成したファイルの改行コードをPerlプログラム内で処理するTipsをいただきました。(2014/09/24)
- 2014年10月04日にHPCIワークショップ「医療とビッグデータ解析」(9:00-9:20)に引き続いて 中級者向けバイオインフォマティクス入門講習会@仙台国際センター(10:50-12:20)で話します。 スライド中のhogeフォルダの圧縮ファイルはhoge.zip(20140929, 22:27版)です。 20140819版から、htmlのスタイルファイル情報を追加して見栄えをよくしただけです(2014/09/29)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGS速習コース。9/3-4の服部先生の講義資料が9/8に、9/11の河岡先生の講義資料が9/18にアップデートされました。 アンケートを提出していない人は早めにお願いします。(2014/09/20)
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGS速習コースで利用する計算機環境構築する一通りの手順を公開しました。(2014/08/11)
- 日本乳酸菌学会誌のNGS関連連載の第1回分PDFを公開しました。 関連項目はこちら。(2014/08/03)
- 2014年9月1日~12日に「バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ)速習コース」を開催します。 受講申込は締め切りました。(2014/08/01)
- 2014年9月1日~12日に「バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ)速習コース」を東大農で開催します。 受講申込は6/24夕方に締め切りましたが、TA申込み枠はまだ若干余裕があります。 TA申込みが全日程受講申込締め切り後の6/24から7/3朝までできない状態になっていたようで失礼しましたm(_ _)m。7/4の10:00ごろに復旧しております。(2014/07/04)
インストール | について
本ウェブページの2015年3月下旬までの推奨インストール手順Rのインストールと起動は、 数十GB程度のHDD容量を必要とすること、そしてそれに伴いインストールにかかる時間、インストールするパッケージ数の増大に伴うエラー遭遇率の上昇のため、 東大有線LANでもストレスを感じるようになってきました。そのため、 ユーザごとのネットワーク環境にも配慮した様々な選択肢を2015年3月末に提供し、推奨手順を変更しました。 私の環境は、Windows PCは(Windows 10; 64 bit)、Macintosh PCはMacBook Pro (OS X Yosemite ver. 10.10; 64 bit)です。 RStudioというものもありますが、私は使いません。 大人数のハンズオン講義を円滑に進められないからですが、通常の個人利用上は全く問題ないと思われます。 以下は、「R本体の最新版」と「必要最小限プラスアルファ(数GB?!)」の推奨インストール手順をまとめたものです。
- Windows版(R_install_win.pdf; 2018.03.12版)
- Macintosh版(R_install_mac.pdf; 2015.04.03版)
遭遇するかもしれないエラーとその対処法を以下に示しました。とりあえずWindows版のみの提供ですが、 Macintoshでも同じようなことが起こるのかもしれません。(2015.11.12追加)
- Windows版(R_install_troubleshoot.pdf; 2015.11.12版)
インストール | R本体 | 最新版 | Win用
最新版(リリース版のこと)は、下記手順を実行します。インストールが無事完了したら、 デスクトップに「R i386 3.X.Y (32 bitの場合; XやYの数値はバージョンによって異なります)」 または「R x64 3.X.Y (64 bitの場合)」アイコンが作成されます。 私は「R i386 3.X.Y」のアイコンは使わないので、いつもゴミ箱に捨てています。 「R x64 3.X.Y」アイコンのほうのみ利用しています。 尚、エンドユーザには実質的に無縁のものだと思いますが開発版(devel版)というのもあります。
- Rのインストーラを「実行」
- 聞かれるがままに「次へ」などを押してとにかくインストールを完了させる
- 「コントロールパネル」−「デスクトップのカスタマイズ」−「フォルダオプション」−「表示(タブ)」−「詳細設定」のところで、 「登録されている拡張子は表示しない」のチェックを外してください。
インストール | R本体 | 最新版 | Mac用
最新版(リリース版のこと)は、下記手順を実行します。インストールが無事完了したら、 Finderを起動して、左のメニューの「アプリケーション」をクリックすると、Rのアイコンが作成されていることが確認できます。 Win同様、エンドユーザには実質的に無縁のものだと思いますがMacにも開発版(devel版)というのがあります。 R-3.1.3までは、R-3.1.3-marvericks.pkgやR-3.1.3-snowleopard.pkgといった記述になっていましたが、R-3.2.0以降は太字部分が消えてます。
- http://cran.r-project.org/bin/macosx/の「R-3.X.Y.pkg」をクリック。 (XやY中の数値はバージョンによって異なります)
- ダウンロードしたファイルをダブルクリックして、聞かれるがままに「次へ」などを押してとにかくインストールを完了させる
- 「Finder」-「環境設定」-「詳細」タブのところで「すべてのファイル名拡張子を表示」にチェックを入れる。
インストール | R本体 | 過去版 | Win用
昔のバージョンをインストールしたい局面もごく稀にあると思います。 その場合は、ここをクリックして、 任意のバージョンのものをインストールしてください。例えば、2014年10月リリースのver. 3.1.2をインストールしたい場合は、 3.1.2をクリックして、 「Download R 3.1.2 for Windows」 をクリックすれば、後は最新版と同じです。
インストール | R本体 | 過去版 | Mac用
昔のバージョンをインストールしたい局面もごく稀にあると思います。 その場合は、ここをクリックして、 任意のバージョンのものをインストールしてください。例えば、2014年10月リリースのver. 3.1.2をインストールしたい場合は、 ページ下部の「R-3.1.2-marvericks.pkg」 をクリックすれば、後は最新版と同じです。
インストール | Rパッケージ | ほぼ全て(20GB以上?!)
Rパッケージの2大リポジトリであるCRANとBioconductor 中のほぼすべてのパッケージをインストールするやり方です。 パソコンのHDD容量に余裕があって、数千個ものパッケージ(20GB程度分)を数時間以上かけてダウンロードしてインストールできる環境にある方は、 R本体を起動し、以下を「R コンソール画面上」でコピー&ペースト。
1. 基本形:
どこからダウンロードするか?と聞かれます。自分のいる場所から近いサイトを指定しましょう。
install.packages(available.packages()[,1], dependencies=TRUE)#CRAN中にある全てのパッケージをインストール source("http://www.bioconductor.org/biocLite.R")#おまじない biocLite(all_group()) #Bioconductor中にあるほぼ全てのパッケージをインストール biocLite("BSgenome.Athaliana.TAIR.TAIR9", suppressUpdates=TRUE)#シロイヌナズナゲノム biocLite("BSgenome.Celegans.UCSC.ce6", suppressUpdates=TRUE)#線虫ゲノム biocLite("BSgenome.Drerio.UCSC.danRer7", suppressUpdates=TRUE)#ゼブラフィッシュゲノム biocLite("BSgenome.Hsapiens.NCBI.GRCh38", suppressUpdates=TRUE)#ヒトゲノム(GRCh38) biocLite("BSgenome.Hsapiens.UCSC.hg19", suppressUpdates=TRUE)#ヒトゲノム(hg19) biocLite("BSgenome.Mmusculus.UCSC.mm10", suppressUpdates=TRUE)#マウスゲノム(mm10)
2. 東大アグリバイオサーバを利用する場合:
2015年4月24現在、東京大学・弥生キャンパスにあるアグリバイオインフォマティクスの居室でのみ利用可能。 動作確認中。寺田透 先生提供情報。
#CRAN中にある全てのパッケージをインストール options(repos="http://finlandia.iu.a.u-tokyo.ac.jp/CRAN")#利用するリポジトリを指定 install.packages(available.packages()[,1], dependencies=TRUE)#全てのパッケージをインストール #Bioconductor中にあるほぼ全てのパッケージをインストール source("http://finlandia.iu.a.u-tokyo.ac.jp/BioC/biocLite.R")#おまじない biocLite(all_group(), suppressUpdates=TRUE)#Bioconductor中にあるほぼ全てのパッケージをインストール biocLite("BSgenome.Athaliana.TAIR.TAIR9", suppressUpdates=TRUE)#シロイヌナズナゲノム biocLite("BSgenome.Celegans.UCSC.ce6", suppressUpdates=TRUE)#線虫ゲノム biocLite("BSgenome.Drerio.UCSC.danRer7", suppressUpdates=TRUE)#ゼブラフィッシュゲノム biocLite("BSgenome.Hsapiens.NCBI.GRCh38", suppressUpdates=TRUE)#ヒトゲノム(GRCh38) biocLite("BSgenome.Hsapiens.UCSC.hg19", suppressUpdates=TRUE)#ヒトゲノム(hg19) biocLite("BSgenome.Mmusculus.UCSC.mm10", suppressUpdates=TRUE)#マウスゲノム(mm10)
インストール | Rパッケージ | 必要最小限プラスアルファ(数GB?!)
(Rで)塩基配列解析、 (Rで)マイクロアレイデータ解析中で利用するパッケージ、 プラスアルファのパッケージをインストールするやり方です。 Rパッケージの2大リポジトリであるCRANとBioconductor から提供されているパッケージ群のうち、一部のインストールに相当しますので、相当短時間でインストールが完了します。 「options(repos="http://cran.ism.ac.jp/")」が使えなくなっているという指摘を受けたので2016.04.11にコメントアウトしました。
1. R本体を起動
2. CRANから提供されているパッケージ群のインストール
以下を「R コンソール画面上」でコピー&ペースト。 どこからダウンロードするか?と聞かれるので、その場合は自分のいる場所から近いサイトを指定しましょう。
#options(repos="http://cran.ism.ac.jp/")#利用するリポジトリを指定(統計数理研究所の場合) #(Rで)塩基配列解析で主に利用 install.packages("clValid") #Dunn's index計算用のパッケージ。2016.11.03追加 install.packages("openxlsx") #EXCELファイル(.xlsx)を直接読み込むためのパッケージ。2015.11.12追加 install.packages("PoissonSeq") install.packages("rbamtools") #BAM形式ファイルを直接読み込むためのパッケージ。2016.09.14追加 install.packages("samr") #(Rで)マイクロアレイデータ解析でも利用 install.packages("seqinr") #(Rで)マイクロアレイデータ解析でも利用 #(Rで)マイクロアレイデータ解析で利用 install.packages("cclust") install.packages("class") install.packages("e1071") install.packages("GeneCycle") install.packages("gptk") install.packages("GSA") install.packages("mixOmics") install.packages("pvclust") install.packages("RobLoxBioC") #2016.05.10追加 install.packages("som") install.packages("st") install.packages("varSelRF") #アグリバイオの他の講義科目で利用予定 install.packages("ape") install.packages("cluster") install.packages("fields") install.packages("glmnet") #2018.05.11追加 install.packages("KernSmooth") install.packages("mapdata") install.packages("maps") install.packages("MASS") install.packages("MVA") install.packages("qqman") #2018.05.11追加 install.packages("RCurl") #2015.07.24追加 install.packages("rgl") install.packages("rrBLUP") #2018.05.11追加 install.packages("scatterplot3d") install.packages("vcfR") #2018.05.11追加
3. Bioconductorから提供されているパッケージ群のインストール
ゲノム配列パッケージ以外です。
source("http://bioconductor.org/biocLite.R")#おまじない
#(Rで)塩基配列解析で主に利用
biocLite("baySeq", suppressUpdates=TRUE)
biocLite("biomaRt", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用
biocLite("Biostrings", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用
#biocLite("BiSeq", suppressUpdates=TRUE)#2016.07.06にコメントアウト(cosmoとdoMCがなくなったようです)
biocLite("BSgenome", suppressUpdates=TRUE)
biocLite("bsseq", suppressUpdates=TRUE)
biocLite("ChIPpeakAnno", suppressUpdates=TRUE)
biocLite("chipseq", suppressUpdates=TRUE)
biocLite("ChIPseqR", suppressUpdates=TRUE)
biocLite("ChIPsim", suppressUpdates=TRUE)
biocLite("cosmo", suppressUpdates=TRUE)
biocLite("CSAR", suppressUpdates=TRUE)
biocLite("DECIPHER", suppressUpdates=TRUE)#2016.12.29追加
biocLite("DEGseq", suppressUpdates=TRUE)
biocLite("DESeq", suppressUpdates=TRUE)
biocLite("DESeq2", suppressUpdates=TRUE)
biocLite("DiffBind", suppressUpdates=TRUE)
biocLite("doMC", suppressUpdates=TRUE)
biocLite("EBSeq", suppressUpdates=TRUE)
biocLite("EDASeq", suppressUpdates=TRUE)
biocLite("EGSEA", suppressUpdates=TRUE)#2018.06.22追加
biocLite("EGSEAdata", suppressUpdates=TRUE)#2018.06.22追加
biocLite("edgeR", suppressUpdates=TRUE)
biocLite("GenomicAlignments", suppressUpdates=TRUE)
biocLite("GenomicFeatures", suppressUpdates=TRUE)
biocLite("ggplot2", suppressUpdates=TRUE)
biocLite("girafe", suppressUpdates=TRUE)
biocLite("GSEABase", suppressUpdates=TRUE)#2018.06.22場所移動
biocLite("GSVA", suppressUpdates=TRUE) #2018.06.22追加
biocLite("GSVAdata", suppressUpdates=TRUE)#2018.06.22場所移動
biocLite("Heatplus", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用(2015.11.12に追加)
biocLite("impute", suppressUpdates=TRUE)
biocLite("limma", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用
biocLite("maSigPro", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用
biocLite("MBCluster.Seq", suppressUpdates=TRUE)
biocLite("msa", suppressUpdates=TRUE) #2015.12.18追加
biocLite("htSeqTools", suppressUpdates=TRUE)
biocLite("NBPSeq", suppressUpdates=TRUE)
biocLite("pcaMethods", suppressUpdates=TRUE)#2018.05.11追加
biocLite("phyloseq", suppressUpdates=TRUE)
biocLite("PICS", suppressUpdates=TRUE)
biocLite("qrqc", suppressUpdates=TRUE)
biocLite("QuasR", suppressUpdates=TRUE)
biocLite("r3Cseq", suppressUpdates=TRUE)
biocLite("recount", suppressUpdates=TRUE)#2018.06.08追加
biocLite("REDseq", suppressUpdates=TRUE)
biocLite("rGADEM", suppressUpdates=TRUE)
biocLite("rMAT", suppressUpdates=TRUE)
biocLite("Rsamtools", suppressUpdates=TRUE)
biocLite("rtracklayer", suppressUpdates=TRUE)
biocLite("segmentSeq", suppressUpdates=TRUE)
biocLite("SeqGSEA", suppressUpdates=TRUE)
biocLite("seqLogo", suppressUpdates=TRUE)
biocLite("ShortRead", suppressUpdates=TRUE)
biocLite("SplicingGraphs", suppressUpdates=TRUE)
biocLite("SRAdb", suppressUpdates=TRUE)
biocLite("tweeDEseqCountData", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用
biocLite("TCC", suppressUpdates=TRUE) #(Rで)マイクロアレイデータ解析でも利用
biocLite("TxDb.Celegans.UCSC.ce6.ensGene", suppressUpdates=TRUE)
biocLite("TxDb.Hsapiens.UCSC.hg19.knownGene", suppressUpdates=TRUE)
biocLite("TxDb.Hsapiens.UCSC.hg38.knownGene", suppressUpdates=TRUE)
biocLite("TxDb.Mmusculus.UCSC.mm10.knownGene", suppressUpdates=TRUE)
biocLite("TxDb.Rnorvegicus.UCSC.rn5.refGene", suppressUpdates=TRUE)
#biocLite("yeastRNASeq", suppressUpdates=TRUE)
#(Rで)マイクロアレイデータ解析で利用
biocLite("affy", suppressUpdates=TRUE)
biocLite("agilp", suppressUpdates=TRUE)
biocLite("annotate", suppressUpdates=TRUE)
biocLite("ArrayExpress", suppressUpdates=TRUE)
biocLite("beadarray", suppressUpdates=TRUE)
biocLite("BeadDataPackR", suppressUpdates=TRUE)
biocLite("betr", suppressUpdates=TRUE)
biocLite("BHC", suppressUpdates=TRUE)
biocLite("Category", suppressUpdates=TRUE)
biocLite("clusterStab", suppressUpdates=TRUE)
biocLite("DNAcopy", suppressUpdates=TRUE)
biocLite("frma", suppressUpdates=TRUE)
biocLite("gage", suppressUpdates=TRUE)
biocLite("gcrma", suppressUpdates=TRUE)
biocLite("genefilter", suppressUpdates=TRUE)
biocLite("GEOquery", suppressUpdates=TRUE)
biocLite("GLAD", suppressUpdates=TRUE)
biocLite("globaltest", suppressUpdates=TRUE)
biocLite("golubEsets", suppressUpdates=TRUE)
biocLite("GSAR", suppressUpdates=TRUE) #2017.01.27追加
biocLite("hgu133a.db", suppressUpdates=TRUE)
biocLite("hgu133plus2probe", suppressUpdates=TRUE)
biocLite("hgug4112a.db", suppressUpdates=TRUE)
biocLite("illuminaMousev2.db", suppressUpdates=TRUE)
biocLite("lumi", suppressUpdates=TRUE)
biocLite("marray", suppressUpdates=TRUE)
biocLite("inSilicoDb", suppressUpdates=TRUE)
biocLite("Mulcom", suppressUpdates=TRUE)
biocLite("multtest", suppressUpdates=TRUE)
biocLite("OCplus", suppressUpdates=TRUE)
biocLite("org.Hs.eg.db", suppressUpdates=TRUE)
biocLite("pathview", suppressUpdates=TRUE)#2016.07.06にPathviewをpathviewに変更
biocLite("pcot2", suppressUpdates=TRUE)
biocLite("pd.rat230.2", suppressUpdates=TRUE)#2017.02.24追加
biocLite("PGSEA", suppressUpdates=TRUE)
biocLite("plier", suppressUpdates=TRUE)
biocLite("puma", suppressUpdates=TRUE)
biocLite("RankProd", suppressUpdates=TRUE)
biocLite("rat2302.db", suppressUpdates=TRUE)
biocLite("rat2302probe", suppressUpdates=TRUE)
biocLite("safe", suppressUpdates=TRUE)
biocLite("SAGx", suppressUpdates=TRUE)
biocLite("sigPathway", suppressUpdates=TRUE)
biocLite("SpeCond", suppressUpdates=TRUE)
biocLite("SPIA", suppressUpdates=TRUE)
biocLite("topGO", suppressUpdates=TRUE)
biocLite("vsn", suppressUpdates=TRUE)
4. Bioconductorから提供されているパッケージ群のインストール
ゲノム配列パッケージです。一つ一つの容量が尋常でないため、 必要に応じてテキストエディタなどに予めコピペしておき、いらないゲノムパッケージを削除してからお使いください。
source("http://bioconductor.org/biocLite.R")#おまじない
biocLite("BSgenome.Athaliana.TAIR.TAIR9", suppressUpdates=TRUE)#シロイヌナズナゲノム
biocLite("BSgenome.Celegans.UCSC.ce6", suppressUpdates=TRUE)#線虫ゲノム
biocLite("BSgenome.Drerio.UCSC.danRer7", suppressUpdates=TRUE)#ゼブラフィッシュゲノム
biocLite("BSgenome.Hsapiens.NCBI.GRCh38", suppressUpdates=TRUE)#ヒトゲノム(GRCh38)
biocLite("BSgenome.Hsapiens.UCSC.hg19", suppressUpdates=TRUE)#ヒトゲノム(hg19)
biocLite("BSgenome.Mmusculus.UCSC.mm10", suppressUpdates=TRUE)#マウスゲノム(mm10)
インストール | Rパッケージ | 必要最小限プラスアルファ(アグリバイオ居室のみ)
(Rで)塩基配列解析、 (Rで)マイクロアレイデータ解析中で利用するパッケージ、 プラスアルファのパッケージをインストールするやり方です。 Rパッケージの2大リポジトリであるCRANとBioconductor から提供されているパッケージ群のうち、一部のインストールに相当します。 東京大学・弥生キャンパスにあるアグリバイオインフォマティクスの居室で立ち上げているサーバからダウンロードさせるようにしていますので アグリバイオインフォマティクスの居室(および講義室)でのみ利用可能ですが、高速にパッケージのインストールが可能です。 寺田透 先生提供情報。
1. R本体を起動
2. パッケージのインストール:
以下をRコンソール画面上でコピペ
#CRAN中にある全てのパッケージをインストール #options(repos="http://finlandia.iu.a.u-tokyo.ac.jp/CRAN")#利用するリポジトリを指定 options(repos="192.168.144.12/CRAN") #利用するリポジトリを指定 install.packages("PoissonSeq") install.packages("samr") #(Rで)マイクロアレイデータ解析でも利用 install.packages("seqinr") #(Rで)マイクロアレイデータ解析でも利用 #(Rで)マイクロアレイデータ解析で利用 install.packages("cclust") install.packages("class") install.packages("e1071") install.packages("GeneCycle") install.packages("gptk") install.packages("GSA") install.packages("mixOmics") install.packages("pvclust") #install.packages("RobLoxBioC") install.packages("som") install.packages("st") install.packages("varSelRF") #アグリバイオの他の講義科目で利用予定 install.packages("ape") install.packages("cluster") install.packages("fields") install.packages("KernSmooth") install.packages("mapdata") install.packages("maps") install.packages("MASS") install.packages("MVA") install.packages("rgl") install.packages("scatterplot3d") #Bioconductor中にあるほぼ全てのパッケージをインストール source("http://finlandia.iu.a.u-tokyo.ac.jp/BioC/biocLite.R")#おまじない source("192.168.144.12/BioC/biocLite.R")#おまじない biocLite("baySeq", suppressUpdates=TRUE) biocLite("biomaRt", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用 biocLite("Biostrings", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用 biocLite("BiSeq", suppressUpdates=TRUE) biocLite("BSgenome", suppressUpdates=TRUE) biocLite("bsseq", suppressUpdates=TRUE) biocLite("ChIPpeakAnno", suppressUpdates=TRUE) biocLite("chipseq", suppressUpdates=TRUE) biocLite("ChIPseqR", suppressUpdates=TRUE) biocLite("ChIPsim", suppressUpdates=TRUE) biocLite("cosmo", suppressUpdates=TRUE) biocLite("CSAR", suppressUpdates=TRUE) biocLite("DEGseq", suppressUpdates=TRUE) biocLite("DESeq", suppressUpdates=TRUE) biocLite("DESeq2", suppressUpdates=TRUE) biocLite("DiffBind", suppressUpdates=TRUE) biocLite("doMC", suppressUpdates=TRUE) biocLite("EBSeq", suppressUpdates=TRUE) biocLite("EDASeq", suppressUpdates=TRUE) biocLite("edgeR", suppressUpdates=TRUE) biocLite("GenomicAlignments", suppressUpdates=TRUE) biocLite("GenomicFeatures", suppressUpdates=TRUE) biocLite("ggplot2", suppressUpdates=TRUE) biocLite("girafe", suppressUpdates=TRUE) biocLite("impute", suppressUpdates=TRUE) biocLite("limma", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用 biocLite("maSigPro", suppressUpdates=TRUE)#(Rで)マイクロアレイデータ解析でも利用 biocLite("MBCluster.Seq", suppressUpdates=TRUE) biocLite("htSeqTools", suppressUpdates=TRUE) biocLite("NBPSeq", suppressUpdates=TRUE) biocLite("phyloseq", suppressUpdates=TRUE) biocLite("PICS", suppressUpdates=TRUE) biocLite("qrqc", suppressUpdates=TRUE) biocLite("QuasR", suppressUpdates=TRUE) biocLite("r3Cseq", suppressUpdates=TRUE) biocLite("REDseq", suppressUpdates=TRUE) biocLite("rGADEM", suppressUpdates=TRUE) biocLite("rMAT", suppressUpdates=TRUE) biocLite("Rsamtools", suppressUpdates=TRUE) biocLite("rtracklayer", suppressUpdates=TRUE) biocLite("segmentSeq", suppressUpdates=TRUE) biocLite("SeqGSEA", suppressUpdates=TRUE) biocLite("seqLogo", suppressUpdates=TRUE) biocLite("ShortRead", suppressUpdates=TRUE) biocLite("SplicingGraphs", suppressUpdates=TRUE) biocLite("SRAdb", suppressUpdates=TRUE) biocLite("TCC", suppressUpdates=TRUE) #(Rで)マイクロアレイデータ解析でも利用 biocLite("TxDb.Celegans.UCSC.ce6.ensGene", suppressUpdates=TRUE) biocLite("TxDb.Hsapiens.UCSC.hg19.knownGene", suppressUpdates=TRUE) biocLite("TxDb.Hsapiens.UCSC.hg38.knownGene", suppressUpdates=TRUE) biocLite("TxDb.Mmusculus.UCSC.mm10.knownGene", suppressUpdates=TRUE) biocLite("TxDb.Rnorvegicus.UCSC.rn5.refGene", suppressUpdates=TRUE) #biocLite("yeastRNASeq", suppressUpdates=TRUE) biocLite("BSgenome.Athaliana.TAIR.TAIR9", suppressUpdates=TRUE)#シロイヌナズナゲノム biocLite("BSgenome.Celegans.UCSC.ce6", suppressUpdates=TRUE)#線虫ゲノム biocLite("BSgenome.Drerio.UCSC.danRer7", suppressUpdates=TRUE)#ゼブラフィッシュゲノム biocLite("BSgenome.Hsapiens.NCBI.GRCh38", suppressUpdates=TRUE)#ヒトゲノム(GRCh38) biocLite("BSgenome.Hsapiens.UCSC.hg19", suppressUpdates=TRUE)#ヒトゲノム(hg19) biocLite("BSgenome.Mmusculus.UCSC.mm10", suppressUpdates=TRUE)#マウスゲノム(mm10)
インストール | Rパッケージ | 必要最小限(数GB?!)
(Rで)塩基配列解析中で利用するパッケージのみインストールするやり方です。 Rパッケージの2大リポジトリであるCRANとBioconductor から提供されているパッケージ群のうち、ごく一部のインストールに相当しますので、相当短時間でインストールが完了します。
1. R本体を起動
2. CRANから提供されているパッケージ群のインストール
以下を「R コンソール画面上」でコピー&ペースト。 どこからダウンロードするか?と聞かれるので、その場合は自分のいる場所から近いサイトを指定しましょう。
install.packages("samr")
install.packages("seqinr")
3. Bioconductorから提供されているパッケージ群のインストール
ゲノム配列パッケージ以外です。
source("http://bioconductor.org/biocLite.R")#おまじない
biocLite("baySeq", suppressUpdates=TRUE)
biocLite("biomaRt", suppressUpdates=TRUE)
biocLite("Biostrings", suppressUpdates=TRUE)
biocLite("BiSeq", suppressUpdates=TRUE)
biocLite("BSgenome", suppressUpdates=TRUE)
biocLite("bsseq", suppressUpdates=TRUE)
biocLite("ChIPpeakAnno", suppressUpdates=TRUE)
biocLite("chipseq", suppressUpdates=TRUE)
biocLite("ChIPseqR", suppressUpdates=TRUE)
biocLite("ChIPsim", suppressUpdates=TRUE)
biocLite("cosmo", suppressUpdates=TRUE)
biocLite("CSAR", suppressUpdates=TRUE)
biocLite("DEGseq", suppressUpdates=TRUE)
biocLite("DESeq", suppressUpdates=TRUE)
biocLite("DESeq2", suppressUpdates=TRUE)
biocLite("DiffBind", suppressUpdates=TRUE)
biocLite("doMC", suppressUpdates=TRUE)
biocLite("EBSeq", suppressUpdates=TRUE)
biocLite("EDASeq", suppressUpdates=TRUE)
biocLite("edgeR", suppressUpdates=TRUE)
biocLite("GenomicAlignments", suppressUpdates=TRUE)
biocLite("GenomicFeatures", suppressUpdates=TRUE)
biocLite("ggplot2", suppressUpdates=TRUE)
biocLite("girafe", suppressUpdates=TRUE)
biocLite("limma", suppressUpdates=TRUE)
biocLite("maSigPro", suppressUpdates=TRUE)
biocLite("MBCluster.Seq", suppressUpdates=TRUE)
biocLite("htSeqTools", suppressUpdates=TRUE)
biocLite("NBPSeq", suppressUpdates=TRUE)
biocLite("phyloseq", suppressUpdates=TRUE)
biocLite("PICS", suppressUpdates=TRUE)
biocLite("qrqc", suppressUpdates=TRUE)
biocLite("QuasR", suppressUpdates=TRUE)
biocLite("r3Cseq", suppressUpdates=TRUE)
biocLite("REDseq", suppressUpdates=TRUE)
biocLite("rGADEM", suppressUpdates=TRUE)
biocLite("rMAT", suppressUpdates=TRUE)
biocLite("Rsamtools", suppressUpdates=TRUE)
biocLite("rtracklayer", suppressUpdates=TRUE)
biocLite("segmentSeq", suppressUpdates=TRUE)
biocLite("SeqGSEA", suppressUpdates=TRUE)
biocLite("seqLogo", suppressUpdates=TRUE)
biocLite("ShortRead", suppressUpdates=TRUE)
biocLite("SplicingGraphs", suppressUpdates=TRUE)
biocLite("SRAdb", suppressUpdates=TRUE)
biocLite("TCC", suppressUpdates=TRUE)
biocLite("TxDb.Celegans.UCSC.ce6.ensGene", suppressUpdates=TRUE)
biocLite("TxDb.Hsapiens.UCSC.hg19.knownGene", suppressUpdates=TRUE)
biocLite("TxDb.Hsapiens.UCSC.hg38.knownGene", suppressUpdates=TRUE)
biocLite("TxDb.Mmusculus.UCSC.mm10.knownGene", suppressUpdates=TRUE)
biocLite("TxDb.Rnorvegicus.UCSC.rn5.refGene", suppressUpdates=TRUE)
biocLite("yeastRNASeq", suppressUpdates=TRUE)
4. Bioconductorから提供されているパッケージ群のインストール
ゲノム配列パッケージです。一つ一つの容量が尋常でないため、 必要に応じてテキストエディタなどに予めコピペしておき、いらないゲノムパッケージを削除してからお使いください。
source("http://bioconductor.org/biocLite.R")#おまじない
biocLite("BSgenome.Athaliana.TAIR.TAIR9", suppressUpdates=TRUE)#シロイヌナズナゲノム
biocLite("BSgenome.Celegans.UCSC.ce6", suppressUpdates=TRUE)#線虫ゲノム
biocLite("BSgenome.Drerio.UCSC.danRer7", suppressUpdates=TRUE)#ゼブラフィッシュゲノム
biocLite("BSgenome.Hsapiens.NCBI.GRCh38", suppressUpdates=TRUE)#ヒトゲノム(GRCh38)
biocLite("BSgenome.Hsapiens.UCSC.hg19", suppressUpdates=TRUE)#ヒトゲノム(hg19)
biocLite("BSgenome.Mmusculus.UCSC.mm10", suppressUpdates=TRUE)#マウスゲノム(mm10)
インストール | Rパッケージ | 個別
多くのBSgenome系パッケージや TxDb系のパッケージは、「ほぼ全て」の手順ではインストールされません。 理由は、BSgenomeはゲノム配列情報のパッケージなので、ヒトゲノムの様々なバージョン、マウスゲノム、ラットゲノムなどを全部入れると大変なことになるからです。 それでもピンポイントで必要に迫られる局面もあると思いますので、ここではRのパッケージを個別にインストールするやり方を示します。
1. ゼブラフィッシュゲノムのパッケージ(BSgenome.Drerio.UCSC.danRer7)をインストールしたい場合:
400MB程度あります...。
param <- "BSgenome.Drerio.UCSC.danRer7"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
2. TxDb.Rnorvegicus.UCSC.rn5.refGeneパッケージのインストールをしたい場合:
param <- "TxDb.Rnorvegicus.UCSC.rn5.refGene"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
3. TxDb.Hsapiens.UCSC.hg38.knownGeneパッケージのインストールをしたい場合:
param <- "TxDb.Hsapiens.UCSC.hg38.knownGene"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
4. 線虫ゲノムのパッケージ(BSgenome.Celegans.UCSC.ce6)をインストールしたい場合:
20MB程度です。
param <- "BSgenome.Celegans.UCSC.ce6" #パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
5. TxDb.Celegans.UCSC.ce6.ensGeneパッケージのインストールをしたい場合:
param <- "TxDb.Celegans.UCSC.ce6.ensGene"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
6. 大腸菌ゲノムのパッケージ(BSgenome.Ecoli.NCBI.20080805)をインストールしたい場合:
20MB程度です。
param <- "BSgenome.Ecoli.NCBI.20080805"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
7. イヌゲノムのパッケージ(BSgenome.Cfamiliaris.UCSC.canFam3)をインストールしたい場合:
550MB程度です。
param <- "BSgenome.Cfamiliaris.UCSC.canFam3"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
8. ショウジョウバエゲノムのパッケージ(BSgenome.Dmelanogaster.UCSC.dm2)をインストールしたい場合:
30MB程度です。
param <- "BSgenome.Dmelanogaster.UCSC.dm2"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
9. イネゲノムのパッケージ(BSgenome.Osativa.MSU.MSU7)をインストールしたい場合:
100MB程度です。
param <- "BSgenome.Osativa.MSU.MSU7"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
(削除予定)Rのインストールと起動
基本的にはインストール | についてをご覧ください。下記は非推奨です(2015.04.02追加)。
2014年7月31日にアップデートしたWindows用のインストール手順はこちら。 2014年5月14日にアップデートしたMac版のインストール手順こちら(by 孫建強氏)もあります。 注意点は、「Mac OS X のバージョンに関わらず R-3.1.0-snowleopard.pkg をインストールしたほうがよい」です。 また、MacintoshのヒトはChIPXpressData のところでこけることが多いようです(小杉孝嗣 氏 提供情報)。 2015年4月2日現在、上記の状況がどうなっているかは不明ですが、インストール | についてのとおり、 推奨のパッケージインストール手順を「ほぼ全て(20GB以上?!)」から 「インストール | Rパッケージ | 必要最小限プラスアルファ(数GB?!)」 に変更しました。
1. Windows release版のインストールの場合:
- Rのインストーラを「実行」
- 聞かれるがままに「次へ」などを押してとにかくインストールを完了させる
- インストールが無事完了したら、デスクトップに出現する「R3.X.Y (32 bitの場合; XやYの数値はバージョンによって異なります)」 または「R x64 3.X.Y (64 bitの場合)」アイコンをダブルクリックして起動
- 以下を、「R コンソール画面上」でコピー&ペーストする。10GB程度のディスク容量を要しますが一番お手軽です。(どこからダウンロードするか?と聞かれるので、その場合は自分のいる場所から近いサイトを指定)
install.packages(available.packages()[,1], dependencies=TRUE)#CRAN中にある全てのパッケージをインストール source("http://www.bioconductor.org/biocLite.R")#おまじない biocLite(all_group()) #Bioconductor中にある全てのパッケージをインストール biocLite("BSgenome.Athaliana.TAIR.TAIR9", suppressUpdates=TRUE)#Bioconductor中にあるBSgenome.Athaliana.TAIR.TAIR9パッケージをインストール
- 「コントロールパネル」−「デスクトップのカスタマイズ」−「フォルダオプション」−「表示(タブ)」−「詳細設定」のところで、「登録されている拡張子は表示しない」のチェックを外してください。
2. Macintoshのインストールの場合:
特にChIPXpressDataのところでこけたヒト用です。 このパッケージがインストールされないようにするやり方です(孫建強氏 作)。
- http://cran.r-project.org/bin/macosx/の「R-3.X.Y-snowleopard.pkg」をクリック。 (XやY中の数値はバージョンによって異なります)
- ダウンロードしたファイルをダブルクリックして、聞かれるがままに「次へ」などを押してとにかくインストールを完了させる
- インストールが無事完了したら、Finderを起動して、左のメニューの「アプリケーション」をクリックし、Rのアイコンをダブルクリックして起動
- 以下を、「R コンソール画面上」でコピー&ペーストする。10GB程度のディスク容量を要しますが一番お手軽です。(どこからダウンロードするか?と聞かれるので、その場合は自分のいる場所から近いサイトを指定)
param <- "ChIPXpressData" #エラーの原因となっているパッケージ名を指定 #install.packages(available.packages()[,1], dependencies=TRUE)#CRAN中にある全てのパッケージをインストール x <- c("UNINSTALLED_PKG_NAME") #インストールされなかったパッケージ名をxに格納 source("http://www.bioconductor.org/biocLite.R")#おまじない pkg <- all_group() #Bioconductor中にある全てのパッケージ名をpkgに格納 pkg <- pkg[pkg != param] #paramで指定したパッケージを除去した結果をpkgに格納 for (i in pkg){ #pkgの要素数分だけループを回す e <- try(biocLite(i, suppressUpdates=TRUE))#パッケージをインストール if(class(e) == "try-error") x <- c(x, i)#インストールに失敗したパッケージ情報をxにどんどん追加 } biocLite("BSgenome.Athaliana.TAIR.TAIR9", suppressUpdates=TRUE)#Bioconductor中にあるBSgenome.Athaliana.TAIR.TAIR9パッケージをインストール
3. Windows devel版(R-devel)のインストールの場合(advanced userのみ):
孫建強氏作のネットワークが不安定な場合でも一つずつ確実にインストールしていくやり方だが、非常に遅いことが判明したので非推奨になりました。
- Rのインストーラを「実行」
- 聞かれるがままに「次へ」などを押してとにかくインストールを完了させる
- R-develを起動し、以下を、「R コンソール画面上」でコピー&ペーストする。
#CRAN中にある全パッケージをインストール x <- c("UNINSTALLED_PKG_NAME") #インストールされなかったパッケージ名をxに格納 available <- available.packages() #利用可能なパッケージ名をavailableに格納 installed <- installed.packages() #インストールされているパッケージ名をinstalledに格納 for (i in available[,1]){ #availableの要素数分だけループを回す idx <- grep(paste("^", i, "$", sep=""), installed[, 1])#available中のパッケージ名iがinstalled中にあるかどうかを判定 if(length(idx) > 0) { #available中のパッケージ名がinstalled中にあるかどうかを判定(あればTRUE) if(installed[idx,3] == available[i,2]){#バージョンも同じかどうかを判定(同じならTRUE) next #nextは、次のパッケージ名へ、という意味 } } e <- try(install.packages(i, dependencies=TRUE))#まだインストールされていないか、インストールされていても古いバージョンのパッケージをインストール if(class(e) == "try-error") x <- c(x, i)#インストールに失敗したパッケージ情報をxにどんどん追加 } uninstalled_cran <- x #インストールに失敗したパッケージ情報をuninstalled_cranに格納 #Bioconductor中にある全パッケージをインストール x <- c("UNINSTALLED_PKG_NAME") #インストールされなかったパッケージ名をxに格納 source("http://www.bioconductor.org/biocLite.R")#おまじない for (i in all_group()){ #all_group()でみられる要素数分だけループを回す e <- try(biocLite(i, suppressUpdates=TRUE))#パッケージiをインストール if(class(e) == "try-error") x <- c(x, i)#インストールに失敗したパッケージ情報をxにどんどん追加 } uninstalled_bioc <- x #インストールに失敗したパッケージ情報をuninstalled_biocに格納
(削除予定)個別パッケージのインストール
Rのインストールと起動に従って推奨手順通りに行えば、ほとんどのパッケージは自動でインストールされます。 しかし、多くのBSgenome系パッケージや TxDb系のパッケージは自動ではインストールされませんので、個別にインストールする必要があります。 ここでは、それらのパッケージのインストール手順を示します。
1. ゼブラフィッシュゲノムのパッケージ(BSgenome.Drerio.UCSC.danRer7)をインストールしたい場合:
400MB程度あります...。
param <- "BSgenome.Drerio.UCSC.danRer7"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
2. TxDb.Rnorvegicus.UCSC.rn5.refGeneパッケージのインストールをしたい場合:
param <- "TxDb.Rnorvegicus.UCSC.rn5.refGene"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
3. TxDb.Hsapiens.UCSC.hg38.knownGeneパッケージのインストールをしたい場合:
param <- "TxDb.Hsapiens.UCSC.hg38.knownGene"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
4. 線虫ゲノムのパッケージ(BSgenome.Celegans.UCSC.ce6)をインストールしたい場合:
20MB程度です。
param <- "BSgenome.Celegans.UCSC.ce6" #パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
5. TxDb.Celegans.UCSC.ce6.ensGeneパッケージのインストールをしたい場合:
param <- "TxDb.Celegans.UCSC.ce6.ensGene"#パッケージ名を指定 #本番 source("http://bioconductor.org/biocLite.R")#おまじない biocLite(param, suppressUpdates=TRUE) #おまじない
基本的な利用法
以下は、インストール | についてを参考にして必要なパッケージのインストールが完了済みのヒトを対象として、 このウェブページの基本的な利用法を簡単に解説したものです。
- Windows版(R_seq_usersguide_win.pdf; 2015.04.03版)
- Macintosh版(R_seq_usersguide_mac.pdf; 2015.04.03版)
サンプルデータ
- Illumina/36bp/single-end/human (SRA000299) data (Marioni et al., Genome Res., 2008)
- Illumina/36bp/single-end/human (SRA000299) data (Marioni et al., Genome Res., 2008)
- Illumina/36bp/single-end/human (SRA000299) data (Marioni et al., Genome Res., 2008)
-
カウントデータ(SupplementaryTable2_changed.txt)と長さ情報ファイル(ens_gene_46_length.txt)を読み込んで、以下を実行して、「配列長情報を含み、カウント数の和が0より大きい行のみ抽出した結果」です。カウントデータファイル(data_marioni.txt)と配列長情報ファイル(length_marioni.txt)。
in_f1 <- "SupplementaryTable2_changed.txt"#入力ファイル名を指定してin_f1に格納(カウントデータファイル) in_f2 <- "ens_gene_46_length.txt" #入力ファイル名を指定してin_f2に格納(長さ情報ファイル) out_f1 <- "data_marioni.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "length_marioni.txt" #出力ファイル名を指定してout_f2に格納 #入力ファイルの読み込み data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")#in_f1で指定したファイルの読み込み len <- read.table(in_f2, header=TRUE, sep="\t", quote="")#in_f2で指定したファイルの読み込み dim(data) #行数と列数を表示 dim(len) #行数と列数を表示 #本番(共通IDのもののみ抽出) rownames(len) <- len[,1] #行の名前としてIDを与えている common <- intersect(rownames(len), rownames(data))#共通IDをcommonに格納 data <- data[common,] #共通IDのもののみ抽出 len <- len[common,] #共通IDのもののみ抽出 dim(data) #行数と列数を表示 dim(len) #行数と列数を表示 head(data) #確認してるだけです head(len) #確認してるだけです #本番(ゼロカウントデータのフィルタリング) obj <- (rowSums(data) > 0) #条件を満たすかどうかを判定した結果をobjに格納 data <- data[obj,] #objがTRUEとなる要素のみ抽出した結果をdataに格納 len <- len[obj,] #objがTRUEとなる要素のみ抽出した結果をlenに格納 dim(data) #行数と列数を表示 dim(len) #行数と列数を表示 head(data) #確認してるだけです head(len) #確認してるだけです #ファイルに保存 tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 write.table(len, out_f2, sep="\t", append=F, quote=F, row.names=F)#lenの中身を指定したファイル名で保存
- ABI_SOLiD/25-35bp/single-end/mouse (SRA000306; EB vs. ES) data (Cloonan et al., Nat Methods, 2008)
- Illumina/50bp/paired-end/mouse (SRA012213; liver) data (Robertson et al., Nat Methods, 2010)
- Illumina/35bp/single-end/human (SRA010153; MAQC) data (Bullard et al., BMC Bioinformatics, 2010)
- NBPSeqパッケージ(Di et al., SAGMB, 10:art24, 2011)中の ArabidopsisのBiological replicatesデータ(G1群3サンプル vs. G2群3サンプル; Cumbie et al., PLoS One, 2011)です。
- ReCountデータベース(Frazee et al., BMC Bioinformatics, 2011)
- Yeastの二群間比較用データ(2 mutant strains vs. 2 wild-types; technical replicates)
-
上記Yeastの二群間比較用データを用いてGC-content normalizationなどを行う場合に必要な情報
yeast genes (SGD ver. r64)のGC含量(yeastGC_6717.txt)やlength情報(yeastLength_6717.txt)。
R Console画面上で「library(EDASeq)」と打ち込んでエラーが出る場合は、a.をコピペで実行してパッケージをインストールしましょう。
a. EDASeqパッケージのインストール:
source("http://www.bioconductor.org/biocLite.R")#おまじない biocLite("EDASeq") #EDASeqパッケージのインストールb. EDASeqパッケージがインストールされていれば以下のコピペでも取得可能:
library(EDASeq) #パッケージの読み込み data(yeastGC) #yeastRNASeqパッケージ中で提供されているyeastのGC含量情報をロード length(yeastGC) #要素数を表示 head(yeastGC) #最初の数個を表示 data(yeastLength) #yeastRNASeqパッケージ中で提供されているyeastの配列長情報をロード length(yeastLength) #要素数を表示 head(yeastLength) #最初の数個を表示 #それぞれ別々のファイルに保存 tmp <- cbind(names(yeastGC), yeastGC) #yeastGCの「names属性情報」と「GC含量」のベクトルを列方向で結合した結果をtmpに格納 write.table(tmp, "yeastGC_6717.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 tmp <- cbind(names(yeastLength), yeastLength)#yeastLengthの「names属性情報」と「配列長」のベクトルを列方向で結合した結果をtmpに格納 write.table(tmp, "yeastLength_6717.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
「10.」と「11.」のファイルをもとに共通遺伝子(6685個)のみからなるのサブセットにしたファイル:
(data_yeast_common_6685.txt; yeastGC_common_6685.txt; yeastLength_common_6685.txt)
以下のコピペでも上記ファイルを得ることができます。
#必要なパッケージをロード library(yeastRNASeq) #パッケージの読み込み library(EDASeq) #パッケージの読み込み #count dataやGC含量情報(SGD ver. r64)の読み込みとラベル情報の作成 data(geneLevelData) #yeastRNASeqパッケージ中で提供されているカウントデータ(geneLevelData)をロード data(yeastGC) #EDASeqパッケージ中で提供されているyeastのGC含量情報(yeastGC)をロード data(yeastLength) #EDASeqパッケージ中で提供されているyeastの配列長情報(yeastLength)をロード #カウントデータ情報(geneLevelData)とGC含量情報(yeastGC)から共通して存在するサブセットを(同じ遺伝子名の並びで)取得 common <- intersect(rownames(geneLevelData), names(yeastGC))#yeastRNASeqパッケージ中で提供されているデータをロード data <- as.data.frame(geneLevelData[common, ])#6685個の共通遺伝子分のカウントデータ行列をデータフレーム形式でdataに格納 GC <- data.frame(GC = yeastGC[common]) #6685個の共通遺伝子分のGC含量ベクトルをデータフレーム形式でGCに格納 length <- data.frame(Length = yeastLength[common])#6685個の共通遺伝子分の配列長ベクトルをデータフレーム形式でlengthに格納 head(rownames(data)) #行列dataの行名(rownames)情報の最初の数個を表示 head(rownames(GC)) #行列GCの行名(rownames)情報の最初の数個を表示 head(rownames(length)) #行列lengthの行名(rownames)情報の最初の数個を表示 #ファイルに保存 tmp <- cbind(rownames(data), data) #「rownames情報」と「カウントデータ」を列方向で結合した結果をtmpに格納 write.table(tmp, "data_yeast_common_6685.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 tmp <- cbind(rownames(GC), GC) #「rownames情報」と「GC含量情報」を列方向で結合した結果をtmpに格納 write.table(tmp, "yeastGC_common_6685.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 tmp <- cbind(rownames(length), length) #「rownames情報」と「配列長情報」を列方向で結合した結果をtmpに格納 write.table(tmp, "yeastLength_common_6685.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
10,000 genes×6 samplesの「複製あり」タグカウントデータ(data_hypodata_3vs3.txt)
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3」の計6サンプル分からなります。
全10,000遺伝子中の最初の2,000個(gene_1〜gene_2000まで)が発現変動遺伝子(DEG)です。
全2,000 DEGsの内訳:最初の90%分(gene_1〜gene_1800)がG1群で4倍高発現、残りの10%分(gene_1801〜gene_2000)がG2群で4倍高発現
以下のコピペでも(数値は違ってきますが)同じ条件のシミュレーションデータを作成可能です。:library(TCC) #パッケージの読み込み set.seed(1000) #おまじない(同じ乱数になるようにするため) tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.2,#シミュレーションデータの作成 DEG.assign = c(0.9, 0.1), #シミュレーションデータの作成 DEG.foldchange = c(4, 4), #シミュレーションデータの作成 replicates = c(3, 3)) #シミュレーションデータの作成 plotFCPseudocolor(tcc) #シミュレーション条件のpseudo-colorイメージを描画 #ファイルに保存 tmp <- cbind(rownames(tcc$count), tcc$count)#「rownames情報」と「カウントデータ」を列方向で結合した結果をtmpに格納 write.table(tmp, "data_hypodata_3vs3.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
上記のTCCパッケージ中のBiological replicatesを模倣した
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3」の計6サンプルからなるシミュレーションデータから、1列目と4列目のデータを抽出した「複製なし」タグカウントデータ(data_hypodata_1vs1.txt)
よって、「G1_rep1, G2_rep1」の計2サンプル分のみからなります。
以下のコピペでも(数値は違ってきますが)同じ条件のシミュレーションデータを作成可能です。:library(TCC) #パッケージの読み込み set.seed(1000) #おまじない(同じ乱数になるようにするため) tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.2,#シミュレーションデータの作成 DEG.assign = c(0.9, 0.1), #シミュレーションデータの作成 DEG.foldchange = c(4, 4), #シミュレーションデータの作成 replicates = c(1, 1)) #シミュレーションデータの作成 plotFCPseudocolor(tcc) #シミュレーション条件のpseudo-colorイメージを描画 #ファイルに保存 tmp <- cbind(rownames(tcc$count), tcc$count)#「rownames情報」と「カウントデータ」を列方向で結合した結果をtmpに格納 write.table(tmp, "data_hypodata_1vs1.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。 10,000 genes×9 samplesの「複製あり」タグカウントデータ(data_hypodata_3vs3vs3.txt) 「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3, G3_rep1, G3_rep2, G3_rep3」の計9サンプル分からなります。 全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。 全3,000 DEGsの内訳:(1)最初の70%分(gene_1〜gene_2100)がG1群で3倍高発現、(2)次の20%分(gene_2101〜gene_2700)がG2群で10倍高発現、(3)残りの10%分(gene_2701〜gene_3000)がG3群で6倍高発現 以下のコピペでも取得可能です。
out_f <- "data_hypodata_3vs3vs3.txt" #出力ファイル名を指定してout_fに格納 param_replicates <- c(3, 3, 3) #G1, G2, G3群のサンプル数をそれぞれ指定 param_Ngene <- 10000 #全遺伝子数を指定 param_PDEG <- 0.3 #発現変動遺伝子の割合を指定 param_FC <- c(3, 10, 6) #G1, G2, G3群の発現変動の度合い(fold-change)をそれぞれ指定 param_DEGassign <- c(0.7, 0.2, 0.1) #DEGのうちG1, G2, G3群で高発現なものの割合をそれぞれ指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #シミュレーションデータの作成 set.seed(1000) #おまじない(同じ乱数になるようにするため) tcc <- simulateReadCounts(Ngene=param_Ngene,#シミュレーションデータの作成 PDEG=param_PDEG, #シミュレーションデータの作成 DEG.assign=param_DEGassign,#シミュレーションデータの作成 DEG.foldchange=param_FC, #シミュレーションデータの作成 replicates=param_replicates)#シミュレーションデータの作成 plotFCPseudocolor(tcc) #シミュレーション条件のpseudo-colorイメージを描画 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), tcc$count)#「rownames情報」と「カウントデータ」を列方向で結合した結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。 10,000 genes×9 samplesの「複製あり」タグカウントデータ(data_hypodata_2vs4vs3.txt) 「G1_rep1, G1_rep2, G2_rep1, G2_rep2, G2_rep3, G2_rep4, G3_rep1, G3_rep2, G3_rep3」の計9サンプル分からなります。 全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。 全3,000 DEGsの内訳:(1)最初の70%分(gene_1〜gene_2100)がG1群で3倍高発現、(2)次の20%分(gene_2101〜gene_2700)がG2群で10倍高発現、(3)残りの10%分(gene_2701〜gene_3000)がG3群で6倍高発現 以下のコピペでも取得可能です。
library(TCC) #パッケージの読み込み set.seed(1000) #おまじない(同じ乱数になるようにするため) tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,#全遺伝子数とDEGの割合を指定 DEG.assign = c(0.7, 0.2, 0.1),#DEGの内訳(G1が70%, G2が20%, G3が10%)を指定 DEG.foldchange = c(3, 10, 6),#DEGの発現変動度合い(G1が3倍、G2が10倍、G3が6倍)を指定 replicates = c(2, 4, 3)) #各群のサンプル数を指定 plotFCPseudocolor(tcc) #シミュレーション条件のpseudo-colorイメージを描画 #ファイルに保存 tmp <- cbind(rownames(tcc$count), tcc$count)#「rownames情報」と「カウントデータ」を列方向で結合した結果をtmpに格納 write.table(tmp, "data_hypodata_2vs4vs3.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
- Illumina/35bp/single-end/human (SRA000299; kidney vs. liver) data (Marioni et al., Genome Res., 2008)
-
ランダムな塩基配列から生成したリファレンスゲノム配列データ(ref_genome.fa)。 48, 160, 100, 123, 100 bpの配列長をもつ、計5つの塩基配列を生成しています。 description行は"contig"という記述を基本としています。塩基の存在比はAが28%, Cが22%, Gが26%, Tが24%にしています。 set.seed関数を利用し、chr3の配列と同じものをchr5としてコピーして作成したのち、2番目と7番目の塩基置換を行っています。 そのため、実際に指定するのは最初の4つ分の配列長のみです。
out_f <- "ref_genome.fa" #出力ファイル名を指定してout_fに格納 param_len_ref <- c(48, 160, 100, 123) #配列長を指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(28, 22, 26, 24) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_desc <- "chr" #FASTA形式ファイルのdescription行に記述する内容 param4 <- 3 #コピーを作成したい配列番号を指定 param5 <- c(2, 7) #コピー先配列の塩基置換したい位置を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #塩基置換関数の作成 enkichikan <- function(fa, p) { #関数名や引数の作成 t <- substring(fa, p, p) #置換したい位置の塩基を取りだす t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成 substring(fa, p, p) <- t_c #置換 return(fa) #置換後のデータを返す } #本番(配列生成) set.seed(1000) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 hoge <- NULL #hogeというプレースホルダの作成 for(i in 1:length(param_len_ref)){ #length(param_len_ref)で表現される配列数分だけループを回す hoge <- c(hoge, paste(sample(ACGTset, param_len_ref[i], replace=T), collapse=""))#ACGTsetの文字型ベクトルからparam_len_ref[i]回分だけ復元抽出して得られた塩基配列をhogeに格納 } #本番(param4で指定した配列をchr5としてコピーし、param5で指定した位置の塩基をそれぞれ置換) hoge <- c(hoge, hoge[param4]) #param4で指定した配列を追加している hoge[length(param_len_ref)+1] <- enkichikan(hoge[length(param_len_ref)+1], param5[1])#塩基置換 hoge[length(param_len_ref)+1] <- enkichikan(hoge[length(param_len_ref)+1], param5[2])#塩基置換 #本番(DNAStringSet形式に変換) fasta <- DNAStringSet(hoge) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をfastaに格納 names(fasta) <- paste(param_desc, 1:length(hoge), sep="")#description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存 writeXStringSet(fasta, file=out_f, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
上記リファレンスゲノム配列データ(ref_genome.fa)に対してbasic alignerでマッピングする際の動作確認用RNA-seqデータ (sample_RNAseq1.fa)とそのgzip圧縮ファイル(sample_RNAseq1.fa.gz)。 リファレンス配列を読み込んで、list_sub3.txtで与えた部分配列を抽出したものです。 どこに置換を入れているかがわかっているので、basic alignerで許容するミスマッチ数を変えてマップされる or されないの確認ができます。 DNAStringSetオブジェクトを入力として塩基置換を行うDNAString_chartr関数を用いて、最後のリードのみ4番目の塩基にミスマッチを入れています。
in_f1 <- "ref_genome.fa" #入力ファイル名を指定してin_f1に格納(multi-FASTAファイル) in_f2 <- "list_sub3.txt" #入力ファイル名を指定してin_f2に格納(リストファイル) out_f <- "sample_RNAseq1.fa" #出力ファイル名を指定してout_fに格納 param <- 4 #塩基置換したい位置を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #塩基置換関数の作成 DNAString_chartr <- function(fa, p) { #関数名や引数の作成 str_list <- as.character(fa) #文字列に変更 t <- substring(str_list, p, p) #置換したい位置の塩基を取りだす t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成 substring(str_list, p, p) <- t_c #置換 fa_r <- DNAStringSet(str_list) #DNAStringSetオブジェクトに戻す names(fa_r) <- names(fa) #description部分の情報を追加 return(fa_r) #置換後のデータを返す } #入力ファイルの読み込み fasta <- readDNAStringSet(in_f1, format="fasta")#in_f1で指定したファイルの読み込み posi <- read.table(in_f2) #in_f2で指定したファイルの読み込み fasta #確認してるだけです #本番 hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:nrow(posi)){ #length(posi)回だけループを回す obj <- names(fasta) == posi[i,1] #条件を満たすかどうかを判定した結果をobjに格納 hoge <- append(hoge, subseq(fasta[obj], start=posi[i,2], end=posi[i,3]))#subseq関数を用いてobjがTRUEとなるもののみに対して、posi[i,2]とposi[i,3]で与えた範囲に対応する部分配列を抽出した結果をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 fasta #確認してるだけです #後処理(最後のリードのparam番目の塩基に置換を入れている) fasta[nrow(posi)] <- DNAString_chartr(fasta[nrow(posi)], param)#指定した位置の塩基置換を実行した結果をfastaに格納 fasta #確認してるだけです #後処理(description部分の作成) description <- paste(posi[,1], posi[,2], posi[,3], sep="_")#行列posiの各列を"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存 writeXStringSet(fasta, file=out_f, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
上記リファレンスゲノム配列データ(ref_genome.fa)に対してbasic alignerでマッピングする際の動作確認用RNA-seqデータ(sample_RNAseq2.fa)とそのgzip圧縮ファイル(sample_RNAseq2.fa.gz)。
リファレンス配列を読み込んで、list_sub4.txtで与えた部分配列を抽出したものです。 基本的にジャンクションリードがbasic alignerでマップされず、splice-aware alignerでマップされることを示すために作成したものです。
in_f1 <- "ref_genome.fa" #入力ファイル名を指定してin_f1に格納(multi-FASTAファイル) in_f2 <- "list_sub4.txt" #入力ファイル名を指定してin_f2に格納(リストファイル) out_f <- "sample_RNAseq2.fa" #出力ファイル名を指定してout_fに格納 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f1, format="fasta")#in_f1で指定したファイルの読み込み posi <- read.table(in_f2) #in_f2で指定したファイルの読み込み fasta #確認してるだけです #本番 hoge <- NULL #塩基配列用プレースホルダhogeを作成 hoge_d <- NULL #description用プレースホルダhoge_dを作成 for(i in 1:nrow(posi)){ #nrow(posi)回だけループを回す uge <- NULL #ugeを初期化 for(j in 1:(length(posi[i,])/3)){ #ncol(posi)/3回だけループを回す obj <- names(fasta)==posi[i,3*(j-1)+1]#条件を満たすかどうかを判定した結果をobjに格納 uge <- paste(uge, subseq(fasta[obj], #subseq関数を用いてobjがTRUEとなるもののみに対して、 start=posi[i,3*(j-1)+2],#「3*(j-1)+2」列目で指定したstart位置から、 end=posi[i,3*(j-1)+3]),#「3*(j-1)+3」列目で指定したend位置で与えた範囲の部分配列を取得し sep="") #それをugeに連結 } hoge <- append(hoge, uge) #hogeにugeを連結 uge_d <- as.character(posi[i,1]) #uge_dの初期値を与えている for(j in 2:(length(posi[i,]))){ #length(posi[i,])回数分だけループを回す uge_d <- paste(uge_d, as.character(posi[i,j]), sep="_")#description情報の作成 } hoge_d <- append(hoge_d, uge_d) #hoge_dにuge_dを連結 } #後処理(DNAStringSet形式に変換) fasta <- DNAStringSet(hoge) #行列posiの各列を"_"で結合したものをdescriptionに格納 names(fasta) <- hoge_d #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存 writeXStringSet(fasta, file=out_f, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
GTF (General Transfer Format)形式のヒトサンプルファイル(human_annotation_sub.gtf)です。
EnsemblのFTPサイトから得たヒトの遺伝子アノテーションファイル("Homo_sapiens.GRCh37.73.gtf.gz")を ここからダウンロードして得て解凍("Homo_sapiens.GRCh37.73.gtf")したのち、 (解凍後のファイルサイズは500MB、2,268,089行×9列のファイルなので)以下のコピペで、ランダムに50,000行分を非復元抽出して得たファイルです。
in_f <- "Homo_sapiens.GRCh37.73.gtf" #入力ファイル名を指定してin_fに格納(目的のタブ区切りテキストファイル) out_f <- "human_annotation_sub.gtf" #出力ファイル名を指定してout_fに格納 param <- 50000 #(入力ファイルの行数以下の)得たい行数を指定 #入力ファイルの読み込み data <- read.table(in_f, header=FALSE, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #本番 hoge <- sample(1:nrow(data), param, replace=F)#入力ファイルの行数からparamで指定した数だけ非復元抽出した結果をhogeに格納 out <- data[sort(hoge),] #hogeで指定した行のみ抽出した結果をoutに格納 dim(out) #オブジェクトoutの行数と列数を表示 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)#outの中身を指定したファイル名で保存
-
GTF (General Transfer Format)形式のヒトサンプルファイル(human_annotation_sub2.gtf)です。
GTFファイル(human_annotation_sub.gtf)の各行の左端に"chr"を挿入したファイルです。
in_f <- "human_annotation_sub.gtf" #入力ファイル名(目的のタブ区切りテキストファイル)を指定してin_fに格納 out_f <- "human_annotation_sub2.gtf" #出力ファイル名を指定してout_fに格納 param <- "chr" #挿入したい文字列を指定 #入力ファイルの読み込み data <- read.table(in_f, header=FALSE, sep="\t", quote="")#in_fで指定したファイルの読み込み #本番(文字列挿入) data[,1] <- paste(param, data[,1], sep="")#dataオブジェクトの1列目の左側にparamで指定した文字列を挿入 #ファイルに保存 write.table(data, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)#dataの中身を指定したファイル名で保存
-
GTF (General Transfer Format)形式のヒトサンプルファイル(human_annotation_sub3.gtf)です。
ヒトゲノム配列("BSgenome.Hsapiens.UCSC.hg19")中の染色体名と一致する遺伝子アノテーション情報のみhuman_annotation_sub2.gtfから抽出したファイルです。
in_f1 <- "human_annotation_sub2.gtf" #入力ファイル名を指定してin_f1に格納(GFF/GTFファイル) in_f2 <- "BSgenome.Hsapiens.UCSC.hg19" #入力ファイル名を指定してin_f2に格納(リファレンス配列) out_f <- "human_annotation_sub3.gtf" #出力ファイル名を指定してout_fに格納 #入力ファイルの読み込み(GFF/GTFファイル) data <- read.table(in_f1, header=FALSE, sep="\t", quote="")#in_f1で指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #前処理(リファレンス配列の染色体名を抽出) param <- in_f2 #paramという名前で取り扱いたいだけです library(param, character.only=T) #paramで指定したパッケージの読み込み tmp <- ls(paste("package", param, sep=":"))#paramで指定したパッケージで利用可能なオブジェクト名を取得した結果をtmpに格納 hoge <- eval(parse(text=tmp)) #文字列tmpをRオブジェクトとしてhogeに格納 keywords <- seqnames(hoge) #染色体名情報を抽出した結果をkeywordsに格納 keywords #確認してるだけです #本番 obj <- is.element(as.character(data[,1]), keywords)#in_f1で読み込んだファイル中の1列目の文字列ベクトル中の各要素がベクトルkeywords中に含まれるか含まれないか(TRUE or FALSE)の情報をobjに格納(集合演算をしている) out <- data[obj,] #objがTRUEとなる行のみ抽出した結果をoutに格納 dim(out) #オブジェクトoutの行数と列数を表示 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)#outの中身をout_fで指定したファイル名で保存
- Illumina/75bp/single-end/human (SRA061145) data (Wang et al., Nucleic Acids Res., 2013)
- Illumina HiSeq 2000/100bp/paired-end/human (GSE42960) data (Chan et al., Hum. Mol. Genet., 2013)
- Illumina Genome Analyzer II/54bp/single-end/human (SRP017142; GSE42212) data (Neyret-Kahn et al., Genome Res., 2013)
- Illumina HiSeq 2000 (GPL14844)/50bp/single-end/Rat (SRP037986; GSE53960) data (Yu et al., Nat Commun., 2014)
- Illumina GAIIx/76bp/paired-end/Drosophila or Illumina HiSeq 2000/100bp/paired-end/Drosophila (SRP009459; GSE33905) data (Graveley et al., Nature, 2011; Brown et al., Nature, 2014)
- Illumina HiSeq 2000/36bp/single-end/Arabidopsis (GSE36469) data (Huang et al., Development, 2012)
- PacBio/xxx bp/Human (ERP003225) data (Sharon et al., Nat Biotechnol., 2013)
- PacBio/xxx bp/Chicken (SRP038897 by DRA; SRP038897 by ENA; SRP038897 by SRA) data (Sharon et al., PLoS One, 2014)
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample32_ref.fastaとsample32_ngs.fasta)です。
50塩基の長さのリファレンス配列を生成したのち、20塩基長の部分配列を10リード分だけランダム抽出したものです。 塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。 リファレンス配列(仮想ゲノム配列)がsample32_ref.fastaで、 10リードからなる仮想NGSデータがsample32_ngs.fastaです。 リード長20塩基で10リードなのでトータル200塩基となり、50塩基からなる元のゲノム配列の4倍シーケンスしていることになります(4X coverageに相当)。 イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample32_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample32_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 50 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 20 #リード長を指定 param_num_ngs <- 10 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample33_ref.fastaとsample33_ngs.fasta)です。
1000塩基の長さのリファレンス配列を生成したのち、20塩基長の部分配列を200リード分だけランダム抽出したものです。 塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。 リファレンス配列(仮想ゲノム配列)がsample33_ref.fastaで、 200リードからなる仮想NGSデータがsample33_ngs.fastaです。 リード長20塩基で200リードなのでトータル4,000塩基となり、1,000塩基からなる元のゲノム配列の4倍シーケンスしていることになります(4X coverageに相当)。 イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample33_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample33_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 1000 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 20 #リード長を指定 param_num_ngs <- 200 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample34_ref.fastaとsample34_ngs.fasta)です。
1000塩基の長さのリファレンス配列を生成したのち、20塩基長の部分配列を500リード分だけランダム抽出したものです。 塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。 リファレンス配列(仮想ゲノム配列)がsample34_ref.fastaで、 500リードからなる仮想NGSデータがsample34_ngs.fastaです。 リード長20塩基で500リードなのでトータル10,000塩基となり、1,000塩基からなる元のゲノム配列の10倍シーケンスしていることになります(10X coverageに相当)。 イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample34_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample34_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 1000 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 20 #リード長を指定 param_num_ngs <- 500 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample35_ref.fastaとsample35_ngs.fasta)です。
10000塩基の長さのリファレンス配列を生成したのち、40塩基長の部分配列を2500リード分だけランダム抽出したものです。 塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。 リファレンス配列(仮想ゲノム配列)がsample35_ref.fastaで、 2500リードからなる仮想NGSデータがsample35_ngs.fastaです。 リード長40塩基で2500リードなのでトータル100,000塩基となり、10,000塩基からなる元のゲノム配列の10倍シーケンスしていることになります(10X coverageに相当)。 イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample35_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample35_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 10000 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 40 #リード長を指定 param_num_ngs <- 2500 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample36_ref.fastaとsample36_ngs.fasta)です。
10000塩基の長さのリファレンス配列を生成したのち、80塩基長の部分配列を5000リード分だけランダム抽出したものです。 塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。 リファレンス配列(仮想ゲノム配列)がsample36_ref.fastaで、 5,000リードからなる仮想NGSデータがsample36_ngs.fastaです。 リード長80塩基で5,000リードなのでトータル400,000塩基となり、10,000塩基からなる元のゲノム配列の40倍シーケンスしていることになります(40X coverageに相当)。 イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample36_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample36_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 10000 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 80 #リード長を指定 param_num_ngs <- 5000 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample37_ref.fastaとsample37_ngs.fasta)です。
10000塩基の長さのリファレンス配列を生成したのち、100塩基長の部分配列を10000リード分だけランダム抽出したものです。 塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。 リファレンス配列(仮想ゲノム配列)がsample37_ref.fastaで、 10,000リードからなる仮想NGSデータがsample37_ngs.fastaです。 リード長100塩基で10,000リードなのでトータル1,000,000塩基となり、10,000塩基からなる元のゲノム配列の100倍シーケンスしていることになります(100X coverageに相当)。 イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample37_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample37_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 10000 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 100 #リード長を指定 param_num_ngs <- 10000 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
- PacBio/xxx bp/Human (SRP036136) data (Tilgner et al., PNAS, 2014)
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ (G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル vs. G4群3サンプル vs. G5群3サンプル)です。
10,000 genes×15 samplesの「複製あり」タグカウントデータ(data_hypodata_3vs3vs3vs3vs3.txt) 「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3, G3_rep1, G3_rep2, G3_rep3, G4_rep1, G4_rep2, G4_rep3, G5_rep1, G5_rep2, G5_rep3」の計15サンプル分からなります。 全10,000遺伝子(Ngene=10000)中の最初の2,000個(gene_1〜gene_2000まで; 20%なのでPDEG=0.2)が発現変動遺伝子(DEG)です。 全2,000 DEGsの内訳:(1)最初の50%分(gene_1〜gene_1000)がG1群で5倍高発現、 (2)次の20%分(gene_1001〜gene_1400)がG2群で10倍高発現、 (3)次の15%分(gene_1401〜gene_1700)がG3群で8倍高発現、 (4)次の10%分(gene_1701〜gene_1900)がG4群で12倍高発現、 (5)残りの5%分(gene_1901〜gene_2000)がG5群で7倍高発現。 以下のコピペでも取得可能です。
library(TCC) #パッケージの読み込み set.seed(1000) #おまじない(同じ乱数になるようにするため) tcc <- simulateReadCounts(Ngene=10000,PDEG=0.2,#全遺伝子数とDEGの割合を指定 DEG.assign=c(0.5,0.2,0.15,0.1,0.05),#DEGの内訳(G1が50%,G2が20%,G3が15%,G4が10%,G5が5%)を指定 DEG.foldchange=c(5,10,8,12,7),#DEGの発現変動度合い(G1が5倍,G2が10倍,G3が8倍,G4が12倍,G5が7倍)を指定 replicates=c(3, 3, 3, 3, 3)) #各群のサンプル数を指定 plotFCPseudocolor(tcc) #シミュレーション条件のpseudo-colorイメージを描画 #ファイルに保存 tmp <- cbind(rownames(tcc$count), tcc$count)#保存したい情報をtmpに格納 write.table(tmp, "data_hypodata_3vs3vs3vs3vs3.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ (G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
10,000 genes×12 samplesの「複製あり」タグカウントデータ(data_hypodata_4vs4vs4.txt) 「G1_rep1, G1_rep2, G1_rep3, G1_rep4, G2_rep1, G2_rep2, G2_rep3, G2_rep4, G3_rep1, G3_rep2, G3_rep3, G3_rep4」の計12サンプル分からなります。 全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。 全3,000 DEGsの内訳:(1)最初の33.3%分(gene_1〜gene_1000)がG1群で5倍高発現、(2)次の33.3%分(gene_1001〜gene_2000)がG2群で5倍高発現、(3)残りの33.3%分(gene_2001〜gene_3000)がG3群で5倍高発現 以下のコピペでも取得可能です。
out_f <- "data_hypodata_4vs4vs4.txt" #出力ファイル名を指定してout_fに格納 param_replicates <- c(4, 4, 4) #G1, G2, G3群のサンプル数をそれぞれ指定 param_Ngene <- 10000 #全遺伝子数を指定 param_PDEG <- 0.3 #発現変動遺伝子の割合を指定 param_FC <- c(5, 5, 5) #G1, G2, G3群の発現変動の度合い(fold-change)をそれぞれ指定 param_DEGassign <- c(1/3, 1/3, 1/3) #DEGのうちG1, G2, G3群で高発現なものの割合をそれぞれ指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #シミュレーションデータの作成 set.seed(1000) #おまじない(同じ乱数になるようにするため) tcc <- simulateReadCounts(Ngene=param_Ngene,#シミュレーションデータの作成 PDEG=param_PDEG, #シミュレーションデータの作成 DEG.assign=param_DEGassign,#シミュレーションデータの作成 DEG.foldchange=param_FC, #シミュレーションデータの作成 replicates=param_replicates)#シミュレーションデータの作成 plotFCPseudocolor(tcc) #シミュレーション条件のpseudo-colorイメージを描画 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), tcc$count)#保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
Blekhman et al., Genome Res., 2010のリアルカウントデータです。 Supplementary Table1で提供されているエクセルファイル(http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls; 約4.3MB) からカウントデータのみ抽出し、きれいに整形しなおしたものがここでの出力ファイルになります。 20,689 genes×36 samplesのカウントデータ(sample_blekhman_36.txt)です。 実験デザインの詳細はFigure S1中に描かれていますが、 ヒト(Homo Sapiens; HS), チンパンジー(Pan troglodytes; PT), アカゲザル(Rhesus macaque; RM)の3種類の生物種の肝臓サンプル(liver sample)の比較を行っています。 生物種ごとにオス3個体メス3個体の計6個体使われており(six individuals; six biological replicates)、 技術的なばらつき(technical variation)を見積もるべく各個体は2つに分割されてデータが取得されています(duplicates; two technical replicates)。 それゆえ、ヒト12サンプル、チンパンジー12サンプル、アカゲザル12サンプルの計36サンプル分のデータということになります。 以下で行っていることはカウントデータの列のみ「ヒトのメス(HSF1, HSF2, HSF3)」, 「ヒトのオス(HSM1, HSM2, HSM3)」,「チンパンジーのメス(PTF1, PTF2, PTF3)」, 「チンパンジーのオス(PTM1, PTM2, PTM3)」, 「アカゲザルのメス(RMF1, RMF2, RMF3)」, 「アカゲザルのオス(RMM1, RMM2, RMM3)」の順番で並び替えたものをファイルに保存しています。 もう少し美しくやることも原理的には可能ですが、そこは本質的な部分ではありませんので、ここではアドホック(その場しのぎ、の意味)な手順で行っています。 当然ながら、エクセルなどでファイルの中身を眺めて完全に列名を把握しているという前提です。 尚、"R1L4.HSF1"と"R4L2.HSF1"が「HSF1というヒトのメス一個体のtechnical replicates」であることは列名や文脈から読み解けます。
#in_f <- "http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls"#入力ファイル名を指定してin_fに格納 in_f <- "suppTable1.xls" #入力ファイル名を指定してin_fに格納 out_f <- "sample_blekhman_36.txt" #出力ファイル名を指定してout_fに格納 #入力ファイルの読み込み hoge <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(hoge) #行数と列数を表示 #サブセットの取得 data <- cbind( #必要な列名の情報を取得したい列の順番で結合した結果をdataに格納 hoge$R1L4.HSF1, hoge$R4L2.HSF1, hoge$R2L7.HSF2, hoge$R3L2.HSF2, hoge$R8L1.HSF3, hoge$R8L2.HSF3, hoge$R1L1.HSM1, hoge$R5L2.HSM1, hoge$R2L3.HSM2, hoge$R4L8.HSM2, hoge$R3L6.HSM3, hoge$R4L1.HSM3, hoge$R1L2.PTF1, hoge$R4L4.PTF1, hoge$R2L4.PTF2, hoge$R6L6.PTF2, hoge$R3L7.PTF3, hoge$R5L3.PTF3, hoge$R1L6.PTM1, hoge$R3L3.PTM1, hoge$R2L8.PTM2, hoge$R4L6.PTM2, hoge$R6L2.PTM3, hoge$R6L4.PTM3, hoge$R1L7.RMF1, hoge$R5L1.RMF1, hoge$R2L2.RMF2, hoge$R5L8.RMF2, hoge$R3L4.RMF3, hoge$R4L7.RMF3, hoge$R1L3.RMM1, hoge$R3L8.RMM1, hoge$R2L6.RMM2, hoge$R5L4.RMM2, hoge$R3L1.RMM3, hoge$R4L3.RMM3) colnames(data) <- c( #列名を付加 "R1L4.HSF1", "R4L2.HSF1", "R2L7.HSF2", "R3L2.HSF2", "R8L1.HSF3", "R8L2.HSF3", "R1L1.HSM1", "R5L2.HSM1", "R2L3.HSM2", "R4L8.HSM2", "R3L6.HSM3", "R4L1.HSM3", "R1L2.PTF1", "R4L4.PTF1", "R2L4.PTF2", "R6L6.PTF2", "R3L7.PTF3", "R5L3.PTF3", "R1L6.PTM1", "R3L3.PTM1", "R2L8.PTM2", "R4L6.PTM2", "R6L2.PTM3", "R6L4.PTM3", "R1L7.RMF1", "R5L1.RMF1", "R2L2.RMF2", "R5L8.RMF2", "R3L4.RMF3", "R4L7.RMF3", "R1L3.RMM1", "R3L8.RMM1", "R2L6.RMM2", "R5L4.RMM2", "R3L1.RMM3", "R4L3.RMM3") rownames(data)<- rownames(hoge) #行名を付加 dim(data) #行数と列数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
Blekhman et al., Genome Res., 2010のリアルカウントデータです。
1つ前の例題41とは違って、technical replicatesの2列分のデータは足して1列分のデータとしています。 20,689 genes×18 samplesのカウントデータ(sample_blekhman_18.txt)です。
#in_f <- "http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls"#入力ファイル名を指定してin_fに格納 in_f <- "suppTable1.xls" #入力ファイル名を指定してin_fに格納 out_f <- "sample_blekhman_18.txt" #出力ファイル名を指定してout_fに格納 #入力ファイルの読み込み hoge <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(hoge) #行数と列数を表示 #サブセットの取得 data <- cbind( #必要な列名の情報を取得したい列の順番で結合した結果をdataに格納 hoge$R1L4.HSF1 + hoge$R4L2.HSF1, hoge$R2L7.HSF2 + hoge$R3L2.HSF2, hoge$R8L1.HSF3 + hoge$R8L2.HSF3, hoge$R1L1.HSM1 + hoge$R5L2.HSM1, hoge$R2L3.HSM2 + hoge$R4L8.HSM2, hoge$R3L6.HSM3 + hoge$R4L1.HSM3, hoge$R1L2.PTF1 + hoge$R4L4.PTF1, hoge$R2L4.PTF2 + hoge$R6L6.PTF2, hoge$R3L7.PTF3 + hoge$R5L3.PTF3, hoge$R1L6.PTM1 + hoge$R3L3.PTM1, hoge$R2L8.PTM2 + hoge$R4L6.PTM2, hoge$R6L2.PTM3 + hoge$R6L4.PTM3, hoge$R1L7.RMF1 + hoge$R5L1.RMF1, hoge$R2L2.RMF2 + hoge$R5L8.RMF2, hoge$R3L4.RMF3 + hoge$R4L7.RMF3, hoge$R1L3.RMM1 + hoge$R3L8.RMM1, hoge$R2L6.RMM2 + hoge$R5L4.RMM2, hoge$R3L1.RMM3 + hoge$R4L3.RMM3) colnames(data) <- c( #列名を付加 "HSF1", "HSF2", "HSF3", "HSM1", "HSM2", "HSM3", "PTF1", "PTF2", "PTF3", "PTM1", "PTM2", "PTM3", "RMF1", "RMF2", "RMF3", "RMM1", "RMM2", "RMM3") rownames(data)<- rownames(hoge) #行名を付加 dim(data) #行数と列数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
-
TCCパッケージ中のシミュレーションデータ(G1群1サンプル vs. G2群1サンプル vs. G3群1サンプル)です。 10,000 genes×3 samplesの「複製なし」タグカウントデータ(data_hypodata_1vs1vs1.txt) 「G1_rep1, G2_rep1, G3_rep1」の計3サンプル分からなります。 全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。 全3,000 DEGsの内訳:(1)最初の70%分(gene_1〜gene_2100)がG1群で3倍高発現、(2)次の20%分(gene_2101〜gene_2700)がG2群で10倍高発現、 (3)残りの10%分(gene_2701〜gene_3000)がG3群で6倍高発現。 以下のコピペでも取得可能です。
out_f <- "data_hypodata_1vs1vs1.txt" #出力ファイル名を指定してout_fに格納 library(TCC) #パッケージの読み込み set.seed(1000) #おまじない(同じ乱数になるようにするため) tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,#全遺伝子数とDEGの割合を指定 DEG.assign = c(0.7, 0.2, 0.1),#DEGの内訳(G1が70%, G2が20%, G3が10%)を指定 DEG.foldchange = c(3, 10, 6),#DEGの発現変動度合い(G1が3倍、G2が10倍、G3が6倍)を指定 replicates = c(1, 1, 1)) #各群のサンプル数を指定 plotFCPseudocolor(tcc) #シミュレーション条件のpseudo-colorイメージを描画 #ファイルに保存 tmp <- cbind(rownames(tcc$count), tcc$count)#「rownames情報」と「カウントデータ」を列方向で結合した結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
- recount2:Collado-Torres et al., Nat Biotechnol., 2017
- pasillaパッケージ中の複製あり2群間比較用カウントデータです(孫建強氏 提供情報)。
- pasillaパッケージ中の複製なし2群間比較用カウントデータです(孫建強氏 提供情報)。
-
FASTA形式ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa) に対してbasic alignerでマッピングする際の動作確認用RNA-seqデータ(sample_RNAseq4.fa)。 リファレンス配列を読み込んで、list_sub9.txtで与えた部分配列を抽出したものです。 GFF3形式ののアノテーションファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3) を用いてマッピング結果からカウント情報を取得する際に、どの領域にマップされたリードがOKなのかを検証するためのリードファイルです。
in_f1 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"#入力ファイル名を指定してin_f1に格納(multi-FASTAファイル) in_f2 <- "list_sub9.txt" #入力ファイル名を指定してin_f2に格納(リストファイル) out_f <- "sample_RNAseq4.fa" #出力ファイル名を指定してout_fに格納 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f1, format="fasta")#in_f1で指定したファイルの読み込み posi <- read.table(in_f2) #in_f2で指定したファイルの読み込み fasta #確認してるだけです #前処理(description部分をスペースで区切り、分割された中から1番目の要素で置き換える) hoge <- strsplit(names(fasta), " ", fixed=TRUE)#names(fasta)中の文字列を" "で区切った結果をリスト形式でhogeに格納 hoge2 <- unlist(lapply(hoge, "[[", 1)) #hogeのリスト中の1番目の要素を抽出してhoge2に格納 names(fasta) <- hoge2 #names(fasta)の中身をhoge2で置換 fasta #確認してるだけです #本番 hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:nrow(posi)){ #length(posi)回だけループを回す obj <- names(fasta) == posi[i,1] #条件を満たすかどうかを判定した結果をobjに格納 hoge <- append(hoge, subseq(fasta[obj], start=posi[i,2], end=posi[i,3]))#subseq関数を用いてobjがTRUEとなるもののみに対して、posi[i,2]とposi[i,3]で与えた範囲に対応する部分配列を抽出した結果をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 fasta #確認してるだけです #後処理(description部分の作成) description <- paste(posi[,1], posi[,2], posi[,3], sep="_")#行列posiの各列を"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存 writeXStringSet(fasta, file=out_f, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
「Kidney 7 samples vs Liver 7 samples」のRNA-seqの遺伝子発現行列データ(SupplementaryTable2.txt)です。 サンプルは二つの濃度(1.5 pM and 3 pM)でシーケンスされており、「3 pMのものが5 samples vs. 5 samples」、「1.5 pMのものが2 samples vs. 2 samples」という構成です。 SupplementaryTable2.txtをエクセルで開くと、7列目以降に発現データがあることがわかります。詳細な情報は以下の通りです(原著論文中のFigure 1からもわかります):
7列目:R1L1Kidney (3 pM) 8列目:R1L2Liver (3 pM) 9列目:R1L3Kidney (3 pM) 10列目:R1L4Liver (3 pM) 11列目:R1L6Liver (3 pM) 12列目:R1L7Kidney (3 pM) 13列目:R1L8Liver (3 pM) 14列目:R2L1Liver (1.5 pM) 15列目:R2L2Kidney (3 pM) 16列目:R2L3Liver (3 pM) 17列目:R2L4Kidney (1.5 pM) 18列目:R2L6Kidney (3 pM) 19列目:R2L7Liver (1.5 pM) 20列目:R2L8Kidney (1.5 pM)
Supplementary table 2のデータを取り扱いやすく加工したデータです。 オリジナルのものは最初の6列が発現データ以外のものだったり、7列目以降も二種類のサンプルが交互に出てくるなど若干R上で表現しずらかったため、以下のようにわかりやすくしたものです。 つまり、サンプルを3pMのものだけにして、「1列目:Genename, 2-6列目:Kidney群, 7-11列目:Liver群」と変更したSupplementaryTable2_changed.txtです:
2列目:R1L1Kidney (3 pM) 3列目:R1L3Kidney (3 pM) 4列目:R1L7Kidney (3 pM) 5列目:R2L2Kidney (3 pM) 6列目:R2L6Kidney (3 pM) 7列目:R1L2Liver (3 pM) 8列目:R1L4Liver (3 pM) 9列目:R1L6Liver (3 pM) 10列目:R1L8Liver (3 pM) 11列目:R2L3Liver (3 pM)
上記SupplementaryTable2_changed.txtをさらに加工したデータ。 NGSデータは(マイクロアレイの黎明期と同じく)金がかかりますので(technical and/or biological) replicatesを簡単には増やせませんので、「1サンプル vs. 1サンプル」比較の局面がまだまだあろうかと思います。 そこで、上記ファイルの2-6列目と7-11列目をそれぞれまとめた(総和をとった)ものSupplementaryTable2_changed2.txtです。
SRR037439から得られるFASTQファイルの最初の2000行分を抽出したMAQC2 brainデータ
非圧縮版:SRR037439.fastq
gzip圧縮版:SRR037439.fastq.gz
26,221 genes×6 samplesの「複製あり」タグカウントデータ(data_arab.txt)
オリジナルは"AT4G32850"というIDのものが重複して存在していたため、19520行目のデータを除去してタブ区切りテキストファイルにしています。
マッピング済みの遺伝子発現行列形式のデータセットを多数提供しています。
7065行 × 4列のyeast RNA-seqデータ(data_yeast_7065.txt)
R Console画面上で「library(yeastRNASeq)」と打ち込んでエラーが出る場合は、a.をコピペで実行してパッケージをインストールしましょう。 このパッケージ中には他に、fastq形式ファイル(500,000 reads)と、bowtieでマッピング(unique mapping and allowing up to two mismatches)したBowtie形式ファイルがそれぞれ4ファイルずつあります。
a. yeastRNASeqパッケージのインストール:
source("http://www.bioconductor.org/biocLite.R")#おまじない
biocLite("yeastRNASeq") #yeastRNASeqパッケージのインストール
b. yeastRNASeqパッケージがインストールされていれば以下のコピペでも取得可能:
library(yeastRNASeq) #パッケージの読み込み data(geneLevelData) #yeastRNASeqパッケージ中で提供されているデータをロード dim(geneLevelData) #行数と列数を表示 head(geneLevelData) #最初の数行を表示 #ファイルに保存 tmp <- cbind(rownames(geneLevelData), geneLevelData)#geneLevelDataの「rownames情報(i.e., 遺伝子名)」と「カウントデータ」の行列を列方向で結合した結果をtmpに格納 write.table(tmp, "data_yeast_7065.txt", sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
ヒト肺の3群間比較用データ:normal human bronchial epithelial (HBE) cells, human lung cancer A549, and H1299 cells
ヒトPBMCというサンプルの2群間比較用データ:未処理群2サンプル (FRDA05-UT and FRDA19.UTB) vs. ニコチンアミド処理群2サンプル(FRDA05-NicoとFRDA19.NB)。 原著論文中では、GSE42960のみが示されていますが、 日米欧三極のDB( SRP017580 by SRA; SRP017580 by DRA; SRP017580 by ENA) からも概観できます。
ペアエンドデータのSRR633902_1.fastqを入力として、最初の1000リード分を抽出することで、 SRR633902_1_sub.fastqを作成しています。 writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。 ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR633902_1.fastq.gz" #入力ファイル名を指定してin_fに格納 out_f <- "SRR633902_1_sub.fastq" #出力ファイル名を指定してout_fに格納 param <- 1000 #抽出したいリード数を指定 #必要なパッケージをロード library(ShortRead) #パッケージの読み込み #入力ファイルの読み込み fastq <- readFastq(in_f) #in_fで指定したファイルの読み込み fastq #fastq情報を表示 #本番(サブセットの抽出) fastq <- fastq[1:param] #サブセットを抽出 fastq #fastq情報を表示 #ファイルに保存 writeFastq(fastq, out_f, compress=F) #fastqの中身を指定したファイル名で保存
ヒトfibroblastsの2群間比較用データ:3 proliferative samples vs. 3 Ras samples
ラットの10組織×雌雄(2種類)×4種類の週齢(2, 6, 21, 104 weeks)×4 biological replicatesの計320サンプルからなるデータ。
ショウジョウバエの様々な組織のデータ(modENCODE)。29 dissected tissue samplesのstrand-specific, paired-endのbiological replicates (duplicates)だそうです。
シロイヌナズナの2群間比較用データ:4 DEX-treated vs. 4 mock-treated
原著論文中では、GSE36469のみが示されていますが、 日米欧三極のDB( SRP011435 by SRA; SRP011435 by DRA; SRP011435 by ENA) からも概観できます。
ヒトの長鎖RNA-seqデータです。配列長はリードによって異なります。
ニワトリの長鎖RNA-seqデータです。配列長はリードによって異なります。
ヒトの長鎖RNA-seqデータです。配列長はリードによって異なります。
ReCount(Frazee et al., BMC Bioinformatics, 2011)の後継版です。 Bioconductor上でもrecountというRパッケージが提供されています。
14,599 genes×7 samplesの「複製あり」タグカウントデータ(sample_pasilla_4vs3.txt)です。 処理前4サンプル(4 untreated) vs. 処理後3サンプル(3 treated)の2群間比較用です。 データの原著論文はBrooks et al., Genome Res., 2011です。 手順としては、pasillaパッケージ中のタブ区切りテキストファイルpasilla_gene_counts.tsvを呼び出し、 それをsample_pasilla_4vs3.txtというファイル名で保存しているだけです。 以下のコピペでも取得可能です。
out_f <- "sample_pasilla_4vs3.txt" #出力ファイル名を指定してout_fに格納 library(pasilla) #パッケージの読み込み #本番 hoge <- system.file("extdata", #pasillaパッケージ中の目的ファイルのフルパス情報を取得 "pasilla_gene_counts.tsv", #pasillaパッケージ中の目的ファイルのフルパス情報を取得 package="pasilla", mustWork=TRUE)#pasillaパッケージ中の目的ファイルのフルパス情報を取得 data <- read.csv(hoge, sep="\t", row.names="gene_id")#gene_id列の情報を行名部分としてread.csv関数で読み込む #ファイルに保存 tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
14,599 genes×2 samplesの「複製なし」タグカウントデータ(sample_pasilla_1vs1.txt)です。 1つ上の例題の4 untreated vs. 3 treatedのオリジナルデータから、1列目と5列目の情報を抽出して、sample_pasilla_1vs1.txtというファイル名で保存しているだけです。 以下のコピペでも取得可能です。
out_f <- "sample_pasilla_1vs1.txt" #出力ファイル名を指定してout_fに格納 param_subset <- c(1, 5) #取り扱いたいサブセット情報を指定 library(pasilla) #パッケージの読み込み #本番 hoge <- system.file("extdata", #pasillaパッケージ中の目的ファイルのフルパス情報を取得 "pasilla_gene_counts.tsv", #pasillaパッケージ中の目的ファイルのフルパス情報を取得 package="pasilla", mustWork=TRUE)#pasillaパッケージ中の目的ファイルのフルパス情報を取得 data <- read.csv(hoge, sep="\t", row.names="gene_id")#gene_id列の情報を行名部分としてread.csv関数で読み込む data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 #ファイルに保存 tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2017
平成29年度NGSハンズオン講習会の門田担当分のコピペ用コード集はここで示します。
- スライドPDF(2017.08.04版; 約14MB)
- プラスアルファの内容からなる補足資料PDF(2017.08.08版; 約3MB)
- W9-2:結果を眺める (スライド6)
- Tips:名前の変更 (スライド8-11)
- Bio-Linux:Field et al., Nat Biotechnol., 2006 (スライド12)
- W9-3:HGAP (Chin et al., Nat Methods, 2013)実行結果ファイルのダウンロード (スライド19-28)
- W9-4:解凍して概観 (スライド32-35)
- 乳酸菌NGS連載第7回のPDFのp104右下 (スライド36)
- W9-5:コンティグ数や行数 (スライド37)
- W10-2のイントロ1 (スライド38)
- W10-2のイントロ2 (スライド39)
- W10-2のイントロ3 (スライド40-41)
- W10-2のイントロ4 (スライド42)
- W10-2のイントロ5 (スライド43)
- W10-2の本番1 (スライド44-46)
- W10-2の本番2 (スライド47)
- W10-2の本番3 (スライド48)
- W10-2の本番4 (スライド49-54)
- 乳酸菌NGS連載第7回のPDFのp106左上 (スライド55)
- W10-3:ファイル分割2 (スライド56)
- W10-4:ファイル分割2 (スライド57-58)
- W10-5:lsで確認 (スライド59-60)
- 乳酸菌NGS連載第7回のPDFのp106左上 (スライド62)
- W11-3のイントロ (スライド63-64)
- W11-3:FASTQ分割1 (スライド65-66)
- W11-3:FASTQ分割2 (スライド67)
- NBDCの平成28年度NGSハンズオン講習会 (スライド69)
- W11-4:スコア分布 (スライド70-76)
- W11-7:pngファイル (スライド77)
- W11-8:テキストファイル (スライド78-79)
- W11-9:sequence3.fq (スライド80)
- less sequence3.txt (スライド82-87)
- 乳酸菌NGS連載第7回のPDFのp106左中 (スライド88)
- 乳酸菌NGS連載第7回のPDFのp106左下 (スライド90)
- W12-4:入力ファイル作成 (スライド91-93)
- W12-5:dotPlot実行 (スライド94-95)
- W12-7:ミニ環状コンティグ作成法 (スライド99)
- W12-8:dotPlot実行 (スライド100)
- W13-2:cutコマンド (スライド106)
- W13-3:続cutコマンド (スライド107-108)
- 乳酸菌NGS連載第7回のPDFのp106右下 (スライド110)
- W14:dotter (Sonnhammer and Durbin, Gene, 1995) (スライド111-121)
- 乳酸菌NGS連載第7回のPDFのp107左上 (スライド124)
- BLAST(本当の原著論文):Altschul et al., J Mol Biol., 1990
- BLAST(Bio-Linuxのblastnで指定されている論文):Zhang et al., J Comput Biol., 2000
- W15-1:makeblastdb (スライド125-128)
- W15-2:blastn (スライド129-130)
- W15-3:結果を眺める (スライド132-136)
- W15-4:ヒット数 (スライド137-138)
- W15-5:grep -n (スライド139)
- W15-6:grep -A (スライド140-141)
- W15-7:less (スライド143-154)
- 乳酸菌NGS連載第7回のPDFのp107右中 (スライド155)
- W16-3:トリム実行 (スライド156-157)
- W16-4:moreで確認 (スライド158-160)
- W17-1:FASTQのトリム (スライド162)
- W17-2:クオリティスコア分布 (スライド163-165)
- W17-3:ドットプロット (スライド166)
- 乳酸菌NGS連載第8回のPDFのp188左中 (スライド169)
- W4:DFAST (スライド170-192)
- 乳酸菌NGS連載第8回のPDFのp188左中 (スライド193)
- W5-2:sequence4.fa同士でdotterを実行 (スライド195-196)
- 乳酸菌NGS連載第8回のPDFのp188右下 (スライド197と199)
- W6-1:makeblastdb (スライド200)
- W6-2:blastn (スライド201)
- ヒット数 (スライド202)
- 乳酸菌NGS連載第8回のPDFのp189左上 (スライド204)
- W7-2:BlastViewer (スライド205)
- W7-3:BLAST再実行 (スライド210)
- W7-5:解釈 (スライド216-226)
- W7-6:grep (スライド228-)
- 乳酸菌NGS連載第8回のPDFのp189左上 (スライド236)
- W8-2:seq1 vs. seq4 (スライド246-247)
- W11-2:ドラフト配列作成 (スライド273)
- W3-0:ファイル分割1 (スライド280)
- W3-0:ファイル分割2 (スライド281)
- W3-1:シェルスクリプト (スライド282-284)
- W3-4:基本形 (スライド289)
- W3-5:解説 (スライド290-292)
- W3-6:echoで確認 (スライド293)
- W3-7:head部分 (スライド294-296)
- W3-8:発展形 (スライド297-302)
- W3-9:echoで確認 (スライド303)
- W3-10:発展形2 (スライド304-305)
- W3-11:実行 (スライド306)
- W3-12:確認 (スライド307-308)
- W3-13:発展形3 (スライド309)
DDBJ Read Annotation Pipeline (DDBJ Pipeline; Nagasaki et al., DNA Res., 2013)実行結果ですが、スライドの画面そのものには行けません。
平成26年度NGSハンズオン講習会の「事前準備 | Bio-Linux 8とRのインストール状況確認(2016.07.19)」
の一番下にある「8.VirtualBox関連Tips(2016.07.04版)」
とほぼ同じ内容です。
result.zip (約13MB)
cd ~/Desktop/mac_share
pwd
wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/DRR054113/result.zip
#cp ~/backup/result.zip .
ls -l
md5sum result.zip
cd ~/Desktop/mac_share
pwd
ls -l result*
unzip result.zip
ls -l result*
ls -ld result*
最終結果ファイル(polished_assembly.fasta)のコンティグ数、description行情報、および行数を表示。
pwd
cd result
pwd
ls -l
grep -c ">" polished_assembly.fasta
grep ">" polished_assembly.fasta
wc polished_assembly.fasta
cdでホームディレクトリに移動し、lsで確認。これから作成予定のbinというディレクトリがないことを確認しているだけ。
pwd
cd
pwd
ls
binというディレクトリを作成し、そこに移動。第6回W12-1とほぼ同じ。
mkdir bin ls cd bin pwd
multi-FASTA形式ファイルを入力として、指定した配列長未満の配列を除き、配列の長い順にソートして返すPythonプログラム (fastaLengthFilter.py; 谷澤靖洋氏作)をダウンロード。 これを流用して0塩基未満と指定して実行することで、配列長順にソートするだけの目的で利用する。 第6回W12-1とほぼ同じ。
clear
pwd
wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/fastaLengthFilter.py
#cp ~/backup/fastaLengthFilter.py .
ls -l
lsで確認し、headで最初の10行分を表示。
pwd
ls -l
head fastaLengthFilter.py
私はパーミッションのところが「rwxr-xr-x」になってなかったら、無条件でchmod 755をやります。第4回W3-1にも解説あり。
pwd
ls -l
chmod 755 fastaLengthFilter.py
ls -l
fastaLengthFilter.pyは配列長の長い順にソートし、description行部分を"sequence1", "sequence2"などと自動変換するので、 0 bp未満のフィルタリングを行うという一見トリッキーなことを行って、フィルタリング以外の機能を利用している。 ~/bin/fastaLengthFilter.pyのように書くことで、~/binに「パスを通す」作業をしていなくても実行できる。 パスについては、第4回のW9-5やW15-5、第6回のW12-3などにも情報あり。
cd ~/Desktop/mac_share/result pwd ls ~/bin/fastaLengthFilter.py polished_assembly.fasta 0 > LH_hgap.fa
fastaLengthFilter.pyの入力ファイルと出力ファイル。 出力ファイルのサイズのほうが入力に比べて若干小さくなっている。この理由は...
pwd
ls -l *.fasta *.fa
multi-FASTAファイルのdescription行部分の記述内容がシンプルになった効果、および...
grep ">" polished_assembly.fasta grep ">" LH_hgap.fa wc LH_hgap.fa
LH_hgap.faのほうは、1行でコンティグの全塩基を記述し、改行が入っていない(この違いが大きく寄与)。 それゆえ「行数 = コンティグ数×2」となり、Linuxコマンドで処理しやすいという特徴を持つ。
wc polished_assembly.fasta wc LH_hgap.fa
sequence1は、最初の2行分に相当する。それゆえ、headコマンドで最初の2行 のみ抽出した結果をsequence1.faというファイル名で保存している。
pwd ls -l LH* head -n 2 LH_hgap.fa > sequence1.fa ls -l sequence1.fa grep ">" sequence1.fa wc sequence1.fa
sequence2と3は、headとtailを組み合わせて目的の配列のみ抽出。連載第3回のW19-3にもあり。sequence4は、tailだけでよい。
pwd ls -l LH* head -n 4 LH_hgap.fa | tail -n 2 > sequence2.fa ls -l sequence2.fa grep ">" sequence2.fa wc sequence2.fa head -n 6 LH_hgap.fa | tail -n 2 > sequence3.fa ls -l sequence3.fa grep ">" sequence3.fa wc sequence3.fa tail -n 2 LH_hgap.fa > sequence4.fa ls -l sequence4.fa grep ">" sequence4.fa wc sequence4.fa
ls -lでファイルサイズを確認。[0-9]は任意の数字1字という意味。
pwd
ls -l sequence*
ls -l sequence[0-9].fa
polished_assembly.fastqのファイルサイズがpolished_assembly.fastaの約2倍で安心。 行数が16行であることから、1コンティグ4行で取り出せると閃く。
cd ~/Desktop/mac_share/result
pwd
ls -l
wc LH* poli*
W10-3やW10-4のファイル分割のやり方と若干違うのは、このような記述の仕方でもOKであることを示すとともに、 シェルスクリプト利用のイントロ(後述のスライド280~)。
pwd ls -l polished_assembly.fast* head -n 4 polished_assembly.fastq | tail -n 4 > sequence1.fq head -n 8 polished_assembly.fastq | tail -n 4 > sequence2.fq head -n 12 polished_assembly.fastq | tail -n 4 > sequence3.fq head -n 16 polished_assembly.fastq | tail -n 4 > sequence4.fq ls -l sequence*
FASTQファイルのdescription部分の行頭は@および+なので、念のため両方で調べている。行数は全て4行。
pwd ls *.fq grep "^@" sequence*.fq grep "^+" sequence*.fq wc sequence*.fq
2016年8月1日の講義資料PDF(約12MB)のスライド100-。
「前処理 | クオリティチェック | PHREDスコアに変換」の例題3を参考にしています。 par(mar=c(4, 4, 0, 0))で、 余白の調整もしています。具体的には、図の下と左側を4行分、それ以外を0行分だけ開けるように指定しています。 pngファイル作成(描画)時にいろいろオプション指定している。 pch=20はプロット時のマーカーを「小さい黒丸」にせよ、 cex=0.5は大きさを通常の0.5倍にせよ、 type="p"は、「点プロット(デフォルト)」にせよ、という意味です。 Windowsのヒトはトリプルクリックで全選択できます。CTRLキーを押しながらワンクリックでも全選択できます(寺田朋子 氏提供情報)。
R -q in_f <- "sequence4.fq" #入力ファイル名を指定してin_fに格納 out_f1 <- "sequence4.png" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sequence4.txt" #出力ファイル名を指定してout_f2に格納 param_fig <- c(700, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(ShortRead) #パッケージの読み込み #入力ファイルの読み込み fastq <- readFastq(in_f) #in_fで指定したファイルの読み込み #本番(PHREDスコアに変換) out <- as(quality(fastq), "matrix") #ASCIIコードのquality scoreをPHRED scoreに変換し、データ構造をmatrixにした結果をoutに格納 colnames(out) <- 1:ncol(out) #列名を付与 rownames(out) <- as.character(id(fastq))#行名を付与 #ファイルに保存(pngファイル) png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 plot(x=1:ncol(out), y=out, pch=20, cex=0.5,#プロット type="p", xlab="position", ylab="PHRED score")#プロット dev.off() #おまじない #ファイルに保存(テキストファイル) tmp <- cbind(colnames(out), as.vector(out))#保存したい情報をtmpに格納 write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F, col.names=F)#tmpの中身を指定したファイル名で保存 q(save="no") pwd ls -l sequence4*
sequence4.png。
Linux上で出力ファイル(sequence4.png)を開く場合は、左側の引き出しアイコンから辿っていくのでもよいし、
「eog sequence4.png&」と打つのでもよい(寺田朋子 氏提供情報)。最後の&ばバックグラウンドジョブ指定(第5回W3-1)。
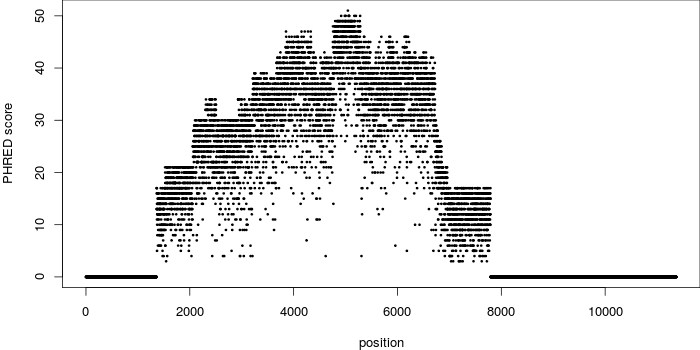{kind=link}
sequence4.txt。最初の5行分、および最後の4行分を表示。
cd ~/Desktop/mac_share/result pwd ls -l sequence4* head -n 5 sequence4.txt tail -n 4 sequence4.txt
W11-4と基本的に同じで入出力のみ異なる。出力ファイルは、sequence3.pngとsequence3.txt。
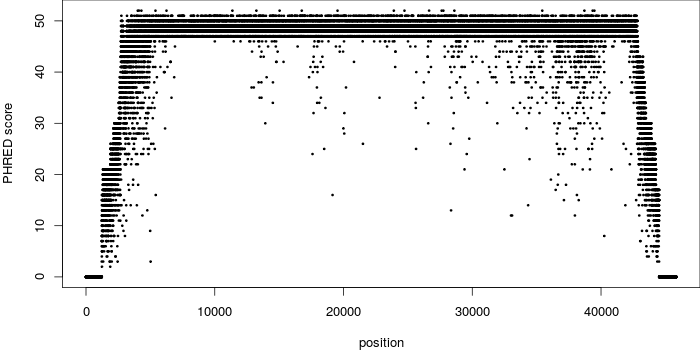{kind=link}
R -q in_f <- "sequence3.fq" #入力ファイル名を指定してin_fに格納 out_f1 <- "sequence3.png" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sequence3.txt" #出力ファイル名を指定してout_f2に格納 param_fig <- c(700, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(ShortRead) #パッケージの読み込み #入力ファイルの読み込み fastq <- readFastq(in_f) #in_fで指定したファイルの読み込み #本番(PHREDスコアに変換) out <- as(quality(fastq), "matrix") #ASCIIコードのquality scoreをPHRED scoreに変換し、データ構造をmatrixにした結果をoutに格納 colnames(out) <- 1:ncol(out) #列名を付与 rownames(out) <- as.character(id(fastq))#行名を付与 #ファイルに保存(pngファイル) png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 plot(x=1:ncol(out), y=out, pch=20, cex=0.5,#プロット type="p", xlab="position", ylab="PHRED score")#プロット dev.off() #おまじない #ファイルに保存(テキストファイル) tmp <- cbind(colnames(out), as.vector(out))#保存したい情報をtmpに格納 write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F, col.names=F)#tmpの中身を指定したファイル名で保存 q(save="no") pwd ls -l sequence3*
cd ~/Desktop/mac_share/result
pwd
ls -l sequence3*
less sequence3.txt
ランダムな塩基配列"ACTCGTCAGA"を含むFASTA形式ファイル(hoge.fa)を作成。 description部分は何でもいいのですが、とりあえず">test"としています。 様々なLinuxテクニックを駆使しています。
cd ~/Desktop/mac_share/result pwd ls -l hoge.fa more hoge.fa echo ">test" echo ">test" > hoge.fa more hoge.fa echo "ACTCGTCAGA" >> hoge.fa more hoge.fa
seqinrパッケージ中のdotPlot関数を用いてドットプロットを作成。 スライド77「eog hoge1.png&」で開くことができます。
cd ~/Desktop/mac_share/result R -q in_f <- "hoge.fa" #入力ファイル名を指定してin_fに格納 out_f <- "hoge1.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(300, 300) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(seqinr) #パッケージの読み込み #入力ファイルの読み込み hoge <- read.fasta(in_f, seqtype="DNA")#in_fで指定したファイルの読み込み hoge #確認してるだけです hoge[[1]] #確認してるだけです #ファイルに保存(pngファイル) png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 0, 0, 0)) #下、左、上、右の順で余白(行)を指定 dotPlot(hoge[[1]], hoge[[1]], xlab="", ylab="")#プロット dev.off() #おまじない q(save="no")
両末端の4塩基が同じACTCGTCAGAACTC"からなるhoge2.faを作成。 最初の10塩基分はW12-4で作成したhoge.faと同じです。
cd ~/Desktop/mac_share/result pwd echo ">test" > hoge2.fa echo "ACTCGTCAGAACTC" >> hoge2.fa more hoge2.fa
出力ファイル名やサイズが異なるだけで、W12-5と基本的に同じ。サイズは、W12-5が10塩基で300ピクセルだったので、 1塩基あたり30ピクセル、よって14塩基の場合は420ピクセルとすれば大きさが揃うと思ったが、 実際にはそうではなかったので微調整した結果350ピクセルになりました。
cd ~/Desktop/mac_share/result R -q in_f <- "hoge2.fa" #入力ファイル名を指定してin_fに格納 out_f <- "hoge2.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(350, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(seqinr) #パッケージの読み込み #入力ファイルの読み込み hoge <- read.fasta(in_f, seqtype="DNA")#in_fで指定したファイルの読み込み hoge #確認してるだけです hoge[[1]] #確認してるだけです #ファイルに保存(pngファイル) png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 0, 0, 0)) #下、左、上、右の順で余白(行)を指定 dotPlot(hoge[[1]], hoge[[1]], xlab="", ylab="")#プロット dev.off() #おまじない q(save="no")
-cオプションは、指定した位置の文字x以降(x-)、 文字まで(-x)、文字の範囲(x-y)を指定できる。 ここでは3-12番目の範囲を指定している。 -fオプションもあり、タブ区切りテキストファイルを読み込んで指定した位置の列情報のみ取り出すこともできます。
cd ~/Desktop/mac_share/result pwd more hoge2.fa tail -n 1 hoge2.fa tail -n 1 hoge2.fa | cut -c 3-12
-cオプションで様々な範囲指定。
cd ~/Desktop/mac_share/result pwd tail -n 1 hoge2.fa tail -n 1 hoge2.fa | cut -c -10 tail -n 1 hoge2.fa | cut -c 1-10 tail -n 1 hoge2.fa | cut -c 5- tail -n 1 hoge2.fa | cut -c 5-14
Bio-Linuxにプレインストールされているdotter (Sonnhammer and Durbin, Gene, 1995) を用いてsequence3.faのドットプロットを描画。 サイズ(ピクセル数)を指定して任意の形式ファイルに保存するやり方はわかりません。
cd ~/Desktop/mac_share/result pwd ls -l sequence3* dotter sequence3.fa sequence3.fa
データベース(DB)側のsequence3.faは塩基配列なので「-dbtype nucl」と指定している。 もしアミノ酸配列の場合は「-dbtype prot」と指定すればよい。 -hash_indexはインデックス配列を作成するためのオプション。無条件でつけておくと間違いない。
cd ~/Desktop/mac_share/result
pwd
makeblastdb -version
makeblastdb -h
pwd
ls sequence3.fa*
makeblastdb -in sequence3.fa -dbtype nucl -hash_index
ls sequence3.fa*
query側もDB側もともにsequence3.fa。BLAST実行結果はsequence3_blast.txtというファイル名で保存。 実物はsequence3_blast.txt。
cd ~/Desktop/mac_share/result
pwd
ls sequence3*
blastn -db sequence3.fa -query sequence3.fa -out sequence3_blast.txt
ls sequence3*
BLAST実行結果はsequence3_blast.txtファイルの最初の10行分(デフォルトが10行)と行数を表示。
pwd
ls -l sequence3_blast.txt
head sequence3_blast.txt
wc sequence3_blast.txt
pwd ls -l sequence3_blast.txt grep "Score =" sequence3_blast.txt grep -c "Score =" sequence3_blast.txt
grep -nで検索文字列を含む行番号も表示。これで、目的のヒット順位のもののアラインメント結果を眺めるときに、行番号情報から辿り易くなる。 viなどの高機能エディタを使えれば、行番号指定で一発で辿り着ける(がそこまでは解説しない)。
pwd
ls -l sequence3_blast.txt
grep -n "Score =" sequence3_blast.txt
wc sequence3_blast.txt
grep -Aで検索文字列("Score =")の後ろ3行分を表示。
grep -A 3 "Score =" sequence3_blast.txt
lessコマンドでsequence3_blast.txtを開き、Score =で検索。 less実行時に-Nオプションをつけることで、行番号を表示することもできる。 画面の横幅を広めにとっておいたほうがよい。第3回のW14-6-2に文字列検索のやり方あり。
cd ~/Desktop/mac_share/result
pwd
ls -l sequence3_blast.txt
less sequence3_blast.txt
W16-2までで眺めた結果を基に、[2450, 43422 bp]領域以外をトリムする。 トリム後の配列はsequence3_trimmed.faに保存。W12-7とW13-2のテクニックを利用。
cd ~/Desktop/mac_share/result pwd ls -l sequence3*.fa echo ">sequence3_trimmed" > sequence3_trimmed.fa more sequence3_trimmed.fa tail -n 1 sequence3.fa | cut -c 2450-43422 >> sequence3_trimmed.fa
cd ~/Desktop/mac_share/result
ls -l sequence3*.fa
more sequence3_trimmed.fa
FASTQ形式ファイル(sequence3.fq)の場合は、2行目(塩基配列情報の行)と4行目(クオリティ情報の行)についてのみ同様な操作を行えばよい。 得られるトリム後のFASTQ形式ファイルはsequence3_trimmed.fq。
cd ~/Desktop/mac_share/result pwd ls -l sequence3*.fq wc sequence3.fq head -n 1 sequence3.fq | tail -n 1 > sequence3_trimmed.fq head -n 2 sequence3.fq | tail -n 1 | cut -c 2450-43422 >> sequence3_trimmed.fq head -n 3 sequence3.fq | tail -n 1 >> sequence3_trimmed.fq head -n 4 sequence3.fq | tail -n 1 | cut -c 2450-43422 >> sequence3_trimmed.fq ls -l sequence3*.fq
トリム後のFASTQ形式ファイル(sequence3_trimmed.fq)を入力として、図1aおよびW11-9と同じようなクオリティスコア分布を作成。 出力ファイルは、sequence3_trimmed.pngとsequence3_trimmed.txt。
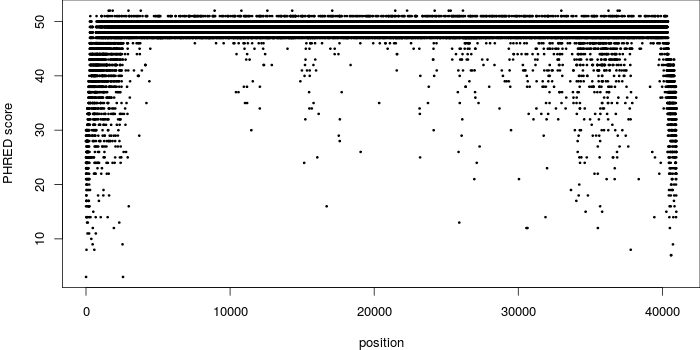{kind=link}
cd ~/Desktop/mac_share/result R -q in_f <- "sequence3_trimmed.fq" #入力ファイル名を指定してin_fに格納 out_f1 <- "sequence3_trimmed.png" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sequence3_trimmed.txt" #出力ファイル名を指定してout_f2に格納 param_fig <- c(700, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(ShortRead) #パッケージの読み込み #入力ファイルの読み込み fastq <- readFastq(in_f) #in_fで指定したファイルの読み込み #本番(PHREDスコアに変換) out <- as(quality(fastq), "matrix") #ASCIIコードのquality scoreをPHRED scoreに変換し、データ構造をmatrixにした結果をoutに格納 colnames(out) <- 1:ncol(out) #列名を付与 rownames(out) <- as.character(id(fastq))#行名を付与 #ファイルに保存(pngファイル) png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 plot(x=1:ncol(out), y=out, pch=20, cex=0.5,#プロット type="p", xlab="position", ylab="PHRED score")#プロット dev.off() #おまじない #ファイルに保存(テキストファイル) tmp <- cbind(colnames(out), as.vector(out))#保存したい情報をtmpに格納 write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F, col.names=F)#tmpの中身を指定したファイル名で保存 q(save="no") pwd ls -l sequence3_trimmed*
dotterでドットプロットを作成して検証。W14でも行った。
cd ~/Desktop/mac_share/result
pwd
ls -l sequence3*.fa
dotter sequence3_trimmed.fa sequence3_trimmed.fa
バクテリア用のアノテーションツールは、他にもMiGAP、
RAST、Prokkaなど多数あります。
より詳細な情報は、「書籍 | 日本乳酸菌学会誌 | 第8回アセンブリ後の解析」のW4-1にあります。
dotter (Sonnhammer and Durbin, Gene, 1995)。得られた結果(ドットプロット)は、[0, 500 bp]と[750-1350]付近の領域が似ていることを意味する。しかし両末端ではないので、 sequence4は環状(プラスミド)ではないのだろうと判断。
cd ~/Desktop/mac_share/result
pwd
ls
dotter sequence4.fa sequence4.fa
BLAST (Zhang et al., J Comput Biol., 2000)実行用のDB側配列のインデックス化。 データベース(DB)側のLH_hgap.faは塩基配列なので「-dbtype nucl」と指定している。 もしアミノ酸配列の場合は「-dbtype prot」と指定すればよい。 -hash_indexはインデックス配列を作成するためのオプション。無条件でつけておくと間違いない。 第7回のW15-1とほぼ同じ。
cd ~/Desktop/mac_share/result
pwd
ls LH_hgap.fa*
makeblastdb -in LH_hgap.fa -dbtype nucl -hash_index
ls LH_hgap.fa*
DB側はLH_hgap.fa、query側はsequence1.fa。BLAST実行結果はsequence1_blast.txtというファイル名で保存。 実物はsequence1_blast.txt。第7回のW15-2とほぼ同じ。
cd ~/Desktop/mac_share/result
pwd
ls -l sequence1*
blastn -db LH_hgap.fa -query sequence1.fa \
-out sequence1_blast.txt
ls -l sequence1*
ls -lh sequence1*
grep -c "Score = "でsequence1_blast.txtのヒット数を表示。 第7回のW15-4とほぼ同じ。
grep -c "Score = " sequence1_blast.txt
Windows版とMacintosh版のみ提供されている。
DB側はLH_hgap.fa、query側はsequence1.fa。BLAST実行結果はXML形式のsequence1_blast.xmlというファイル名で保存。 実物はsequence1_blast.xml。-outfmt 5がXML形式指定に相当。
cd ~/Desktop/mac_share/result
pwd
ls -l sequence1*
blastn -db LH_hgap.fa -query sequence1.fa \
-out sequence1_blast.xml -outfmt 5
ls -lh sequence1*
BlastViewerの結果をsequence1_blast.txtと合わせて確認。
cd ~/Desktop/mac_share/result
pwd
ls -l sequence1*
less sequence1_blast.txt
grepでsequence1_blast.txtの結果の概要を把握。 -nで"Score = "と一致した行番号情報も表示。 -A 3で一致した行を含め後ろの3行分を表示。
cd ~/Desktop/mac_share/result pwd ls -l sequence1* grep -c "Score = " sequence1_blast.txt grep "Score = " sequence1_blast.txt | head grep "Score = " sequence1_blast.txt | head -n 3 grep -A 3 "Score = " sequence1_blast.txt | head -n 15 pwd ls -l sequence1* grep -n "> sequence" sequence1_blast.txt grep -n "Score = " sequence1_blast.txt | tail -n 6
grepでsequence1_blast.txtの結果の概要を把握。 -nと-A 3を組み合わせて利用。さらにパイプ(|)でsequence4の検索結果が含まれるギリギリのtail -n 19として、 必要最小限を表示。
cd ~/Desktop/mac_share/result pwd ls -l sequence1* grep -n -A 3 "Score = " sequence1_blast.txt | tail -n 19
LH_hgap.faを入力として、 重複除去を行った結果をLH_draft.faに保存。 第7回W16-3と同じテクニック。
cd ~/Desktop/mac_share/result pwd ls -l LH*.fa echo ">chromosome" > LH_draft.fa head -2 LH_hgap.fa | tail -1 | cut -c 5839-2283819 >> LH_draft.fa echo ">plasmid1" >> LH_draft.fa head -4 LH_hgap.fa | tail -1 | cut -c 2641-84270 >> LH_draft.fa echo ">plasmid2" >> LH_draft.fa head -6 LH_hgap.fa | tail -1 | cut -c 2450-43422 >> LH_draft.fa ls -l LH*.fa
第7回W10ではLH_hgap.faを入力としたが、 ここではLH_draft.faを入力としたやり方で示します。 第8回W3とは若干関係個所が変わっています。
cd ~/Desktop/mac_share/result
pwd
ls -l LH*.fa
grep ">" LH_draft.fa
wc LH_draft.fa
第7回W10-3からW10-4と同じような書き方だと、こんな感じになる。
cd ~/Desktop/mac_share/result pwd ls -l LH*.fa head -n 2 LH_draft.fa > mongee1.fa head -n 4 LH_draft.fa | tail -n 2 > mongee2.fa tail -n 2 LH_draft.fa > mongee3.fa ls -l LH_draft.fa mongee*
第8回W3-1とほぼ同じ。
cd ~/Desktop/mac_share/result pwd ls -l LH*.fa head -n 2 LH_draft.fa | tail -n 2 > contig1.fa head -n 4 LH_draft.fa | tail -n 2 > contig2.fa head -n 6 LH_draft.fa | tail -n 2 > contig3.fa ls -l mongee* contig* diff mongee1.fa contig1.fa diff mongee2.fa contig2.fa diff mongee3.fa contig3.fa
シェルスクリプトでループを回す基本形。JST-NBDC_1.sh
cd ~/Desktop/mac_share/result
pwd
wget -cq http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JST-NBDC_1.sh
#cp ~/backup/JST-NBDC_1.sh .
ls -l *.sh
more JST-NBDC_1.sh
JST-NBDC_1.shの中身は以下と同じです。 出力ファイル名をtest[0-9].faとしています。 目的は、sequence[0-9].faと同じ結果になるかどうかをdiffコマンドで確認することです。
#!/bin/sh for i in `seq 1 3` do echo "head -n $i*2 LH_draft.fa | tail -n 2 > test$i.fa" done
shコマンドでJST-NBDC_1.shの中身を実行。 実際には実行する予定のコマンドをまだechoで表示させているだけ。
cd ~/Desktop/mac_share/result
pwd
ls *.sh
sh JST-NBDC_1.sh
headコマンド実行時に-n 3*2のような書き方をしてはいけません。
head -n 3*2 LH_draft.fa head -n 3*2 LH_draft.fa | tail -n 2 head -n 6 LH_draft.fa | tail -n 2
シェルスクリプトでループを回す発展形。JST-NBDC_2.sh。 スライドでは-cqとしていますが、ダウンロード状況がわかるように-cのみにしています。
cd ~/Desktop/mac_share/result
pwd
wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JST-NBDC_2.sh
#cp ~/backup/JST-NBDC_2.sh .
ls -l *.sh
more JST-NBDC_2.sh
shコマンドでJST-NBDC_2.shの中身を実行。 実際には実行する予定のコマンドをまだechoで表示させているだけ。
cd ~/Desktop/mac_share/result
pwd
ls -l *.sh
sh JST-NBDC_2.sh
スライドでは-cqとしていますが、ダウンロード状況がわかるように-cのみにしています。 動作確認できたので、画面表示ではなく実際に実行するためにechoを外しているだけです。JST-NBDC_3.sh
cd ~/Desktop/mac_share/result
pwd
wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JST-NBDC_3.sh
#cp ~/backup/JST-NBDC_3.sh .
ls -l *.sh
more JST-NBDC_3.sh
shコマンドでJST-NBDC_3.shを実行。
pwd
ls *.fa
sh JST-NBDC_3.sh
ls *.fa
diffコマンドで違いがない(同じ結果になる)ことを確認。
pwd
ls -l contig*.fa test*.fa
diff contig1.fa test1.fa
diff contig2.fa test2.fa
diff contig3.fa test3.fa
diff test2.fa test3.fa
計4回のループ程度だと違いがわかりませんが、超高速版です。 「expr 遅い シェル」などでググるといいでしょう。詳細説明は省略。 JST-NBDC_4.sh
pwd
wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JST-NBDC_4.sh
#cp ~/backup/JST-NBDC_4.sh .
more JST-NBDC_4.sh
sh JST-NBDC_4.sh
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2016
平成28年度NGSハンズオン講習会の実習で用いるリンク先、 データファイル、コピペ用コード集などはここで示します。
- はじめに(講習会参加者必読) (last modified 2016/08/22)
- 事前準備 | Bio-Linux 8とRのインストール状況確認(2016.07.19) (last modified 2016/07/13)
- 第1部 | 統計解析 | について (last modified 2016/07/14)
- 第1部 | 統計解析 | ゲノム解析、塩基配列解析(2016.07.20) (last modified 2016/07/12)
- 第1部 | 統計解析 | トランスクリプトーム解析1(2016.07.21) (last modified 2016/07/12)
- 第1部 | 統計解析 | トランスクリプトーム解析2(2016.07.22) (last modified 2016/07/13)
- 第2部 | NGS解析(初~中級) | について (last modified 2016/08/19)
- 第2部 | NGS解析(初~中級) | NGS解析基礎(2016.07.25) (last modified 2016/08/03)
- 第2部 | NGS解析(初~中級) | ゲノムReseq、変異解析(2016.07.26) (last modified 2016/08/03)
- 第2部 | NGS解析(初~中級) | RNA-seq(2016.07.27) (last modified 2016/08/03)
- 第2部 | NGS解析(初~中級) | ChIP-seq(2016.07.28) (last modified 2016/08/03)
- 第3部 | NGS解析(中~上級) | について (last modified 2016/08/12)
- 第3部 | NGS解析(中~上級) | Linux環境でのデータ解析:JavaやRの利用法(2016.08.01) (last modified 2016/07/31)
- 第3部 | NGS解析(中~上級) | Linux環境でのデータ解析:マッピング、トリミング、アセンブリ(2016.08.02) (last modified 2016/07/31)
- 第3部 | NGS解析(中~上級) | クラウド環境との連携、ロングリードデータの解析(2016.08.03) (last modified 2016/07/06)
- 第3部 | NGS解析(中~上級) | トランスクリプトームアセンブリ、発現量推定(2016.08.04) (last modified 2016/08/12)
はじめに(講習会参加者必読)
ほぼ完成です(2016年07月19日現在)。
- 本講習会は、これまでの講師の先生方やボランティアTAの方々など、数多くの皆様のご協力によって開催されてきましたm(_ _)m。 個人情報?!の関係上、講義資料中には掲載しておりませんが、毎日講義前と講義後にご覧いただければ幸いですm(_ _)m また、このウェブページの中身に関するバグレポートやよりよい情報提供をいただいた、数えきれないほど多くの関係諸氏にも感謝申し上げますm(_ _)m
- 本講習会の案内や予習事項の指示内容など全般についてですが、多少の記載内容の重複、矛盾、
情報のアップデート漏れなどがあるかもしれませんがご了承ください。
- 受講申し込み段階で、いろいろ要望が出ていることは承知しております。しかし3週間弱にわたる講義室の確保だけでも現実にはかなり大変です。
土日は建物がデフォルトで施錠されます。7/29(Fri.)は消防点検か何かで使えません。
2回に分けて1回あたりの受講人数を減らしてはどうか?的な提案も過去にありましたが、講義室、スタッフ、講師の時間の確保、
PCの再セットアップ、そして開催にかかる諸費用など、費用対効果を総合的に勘案しております。
- 人数を限定する案なども頂戴しますが、なるべく受講希望者全員にタイムリーに受講機会を与えるのは重要だと思っています。
世話人(門田)自体が一年契約の雇用形態なので、毎年「今年で最後。受講生に何を遺すか」という気持ちでやっています
(最初の1年のみで、まさか3年連続で自分が担当するとは思ってもいませんでしたが)。
- もちろん過去の受講生の要望は、可能な範囲で取り入れています。最も重視したのは、下記の2点です。 開催側の負担感はハンパないですが、受講生側にも応分の負担(主に事前準備や予習)を要求し、全体として満足度の高い講習会を目指しています。
- (講師数を減らし)項目間の連携を強化
- 部分受講を認める
- 以下の過去の講習会の報告書、および平成28年度の概要を読んでおいてください。過去の受講生がどういう感想や要望をもち、 世話人がそれに対して次年度にどういう対応を行ってきたのか、といったあたりです。
- 平成26年度報告書:h26_ngs_report.pdf(約4MB)。特にp24以降。
- 平成27年度報告書:h27_ngs_report.pdf(約2MB)。特にp3-6、p10-13、p27以降。
- 平成28年度の概要:h28-h27contents.pdf(約2MB)。
NBDCの平成28年度講習会ページからみられる
「*昨年度との違い、対応関係、想定受講者、および予習事項については
こちら(PDF:2.01MB)をご覧ください。」と同じものです。
- 第2部および第3部でBio-Linuxを採用したのは、WindowsユーザもMacintoshユーザもほぼ同じGUI
で統一的に講義を進められるというのが一番の理由です。
ここでのGUIとは、「画面の見栄えや操作感」という解釈で差支えありません。
- 「事前準備 | Bio-Linux 8とRのインストール状況確認(2016.07.19)」に不参加のヒトも、この項目ををよく読んでおきましょう。
- 受講生はお客様ではありません。講師は神様ではありません。世話人は独裁者です。
- 受講希望者数:162名(締切時点) --> 152名(7/19朝)
- 事前準備(2016年07月19日):127名(締切時点) --> 118名(7/19朝)
- 第1部(2016年07月20日):143名(締切時点) --> 133名(7/19朝), 講義資料PDF(2016.07.12版; 約7MB)とhoge.zip(2016.05.13版; 約2MB)
- 第1部(2016年07月21日):139名(締切時点) --> 130名(7/19朝), 講義資料PDF(2016.07.12版; 約7MB)とhoge.zip(2016.05.23版; 約3MB)
- 第1部(2016年07月22日):139名(締切時点) --> 130名(7/19朝), 講義資料PDF(2016.07.13版; 約7MB)とhoge.zip(2016.05.23版; 約1MB)
- アグリバイオ大学院講義科目「農学生命情報科学特論I (特論I)」受講希望者数は49名。
このうち大学院修了に必要な単位取得希望者数(UT-mate登録者数)は12名。
特論Iの課題(kadai_tokuronI.pdf)と仮想NGSデータ(kadai_20160720.fasta)を2016年7月14日に公開しました。
提出期限は8月20日です。
- 第2部(2016年07月25日):138名(締切時点) --> 129名(7/19朝), 講義資料PDF(2016.08.03版), コピペ用コマンド集
- 第2部(2016年07月26日):132名(締切時点) --> 123名(7/19朝), 講義資料PDF(2016.08.03版), コピペ用コマンド集
- 第2部(2016年07月27日):137名(締切時点) --> 128名(7/19朝), 講義資料PDF(2016.08.03版), コピペ用コマンド集
- 第2部(2016年07月28日):123名(締切時点) --> 117名(7/19朝), 講義資料PDF(2016.08.03版), コピペ用コマンド集
- 第3部(2016年08月01日):119名(締切時点) --> 114名(7/19朝), 講義資料PDF(2016.07.31版; 約17MB)
- 第3部(2016年08月02日):122名(締切時点) --> 115名(7/19朝), 講義資料PDF(2016.07.31版; 約10MB)
- 第3部(2016年08月03日):119名(締切時点) --> 112名(7/19朝), 講義資料PDF(2016.07.06版; 約10MB)
- 第3部(2016年08月04日):116名(締切時点) --> 108名(7/19朝), 講義資料PDF(2016.08.12版; 約10MB)
- アグリバイオ大学院講義科目「農学生命情報科学特論II (特論II)」受講希望者数は46名。
このうち大学院修了に必要な単位取得希望者数(UT-mate登録者数)は11名。
特論IIの課題(kadai_tokuronII.pdf)と仮想NGSデータ(kadai_20160720.fasta)を2016年7月14日に公開しました。
提出期限は8月30日です。
- 「受講生はお客様ではない」とも関連しますが、スタッフに対する暴言・文句的な類のものは一切認めません。
第一義的には「運営側の方針に無条件で従う」です。最終的な全責任の所在は、世話人一個人(門田)になります。
- 障害等のため、設備等の配慮が必要な場合は運営に申し出てください。可能な限り配慮させていただきます。
例えば「近視のためスクリーンに近い場所(前のほうの座席)の確保を希望」などを想定しています。
- 持込PCのヒトも貸与PCのヒトも、PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB)を一通り眺めておいてください。
- 以下は自分のための備忘録(暫定版)
- 住所(居住地域または所属機関所在地;24都道府県)
- 研究分野(複数回答可)
- 職種
- 平成26年度のNGS速習コース受講の有無
- 平成27年度のNGSハンズオン講習会受講の有無
- 実習PC環境
acknowledgement_20160704__.pdf(last modified 2016/07/04)
各日の受講希望者数は下記の通り(2016年07月19日朝の速報値)。 当日の実受講者数は多くても110名程度になるのではと予想していましたが、思ったよりもキャンセルが少ないというのが率直な感想です。 連絡なしの不参加も一定数見込まれますので、実受講者数はもう少し減ります。
東京:93名、神奈川:18名、埼玉:9名、千葉:8名、茨城:4名、京都:4名、静岡:3名、栃木:3名、愛知:2名、宮城:2名、大阪:2名、兵庫:2名、 岐阜、熊本、群馬、広島、三重、新潟、石川、長崎、長野、福岡、北海道、和歌山が各1名
生物学(総合生物学を含む):99名、医歯薬学:66名、農学:52名、情報学:14名、理工学:3名、その他:10名
学生(大学院生):84名、ポスドク:13名、講師・助教:16名、教授・准教授:4名、研究員:23名、主任研究員:4名、研究支援員:5名、その他:14名
受講していない:154名、受講した:5名、未選択:4名
受講していない:142名、受講した:18名、未選択:3名
Mac持込:61名、Win持込:50名、貸与希望:48名
事前準備 | Bio-Linux 8とRのインストール状況確認(2016.07.19)
ほぼ完成です(2016年07月14日現在)。基本的に昨年度と同じく自習です。 スタッフが複数人常駐予定ですので、インストールで躓いた箇所の相談など個別対応してもらってください。 持込PCのヒトも貸与PCのヒトも、PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB)を一通り眺めておいてください。
- Bio-Linux8(第2部および3部で利用するovaファイル)の導入確認
- ovaファイルの導入手順:Windows用(2015.12.28版; 約2MB)
- ovaファイルの導入手順:Macintosh用(2015.12.28版; 約2MB)
- 共有フォルダ設定完了確認
- 第2部参加者(Win):「C:\Users\株式会社\Desktop\share」を変更
- 第2部参加者(Mac):「C:\Users\株式会社\Desktop\share」を変更
- 第3部参加者(Win):「C:\Users\iu\Desktop\share」を変更
- 第3部参加者(Mac):「C:\Users\iu\Desktop\share」を変更
- 基本的なLinuxコマンドの習得状況確認
- R本体およびパッケージのインストール確認
- 講師指定の事前予習内容の再確認
- 第1部 | 統計解析 | について (last modified 2016/07/14)
- 第2部 | NGS解析(初~中級) | について (last modified 2016/08/19)
- 第3部 | NGS解析(中~上級) | について (last modified 2016/08/12)
- 講習会期間中に貸与されるノートPCを用いた各種動作確認
- 無線LANの設定(持ち込みPCのみ)
- VirtualBox関連Tips(2016.07.04版)
- BioLinux8の名前を変えたい
- 使用メモリを変更したい
- ...
第2部および3部で利用するovaファイルは6GBから8GB程度になります。そしてそれを使って第2部および3部を受講してもらいます。 ovaファイルは独立に提供しています。 7/8に受講生あてにダウンロード用URLの連絡がいっているはずです(7/11に第2部用ovaファイルの差し替え案内もいっているはず)。 もしまだ連絡が届いていない受講生がいらっしゃいましたらその旨お伝えください。 例えば全日程参加者は、第2部用ovaファイルと第3部用ovaファイルを独立にダウンロードしておいてください。 ネットワーク環境的にダウンロードできないヒトも一定数いると思われます。 当日は、それらの人々用に第2部および3部のovaファイルが入ったUSBメモリを講義室で用意する予定ですので、 下記を参考にして各自のPCにコピーしてBio-Linuxを導入してください(もちろん乳酸菌NGS連載第2回最後の「1. VirtualBox、および2. Extension Pack」のインストールが完了しているという前提です)。
第2部および第3部参加者のみ。PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB)の項目3にも情報があります。
第2部および第3部参加者のみ。
基本的に第1部参加者のみでよい。
基本的に貸与希望者のみではありますが、持込PCの不具合時にはアグリバイオPCを貸し出します。
この際に普段利用するPC環境でないので戸惑うかもしれません。
この日を利用して貸与PCに慣れておくというのもアリでしょう。
会場で利用可能なアクセスポイントの把握とパスワードの設定を行ってください(PWは当日スタッフから教えてもらってください)。
下記内容が含まれます。
第1部 | 統計解析 | について
第1部は、Bio-Linux上のR環境ではなく、Windows(またはMacintosh)上のR環境で行います。 多少変更するかもしれませんが、ほぼ完成です(2016年07月14日現在)。2016年6月14日に、第1部の心構え(掟)を追加しました。 2016年7月14日に「農学生命情報科学特論I」受講者用の 課題(kadai_tokuronI.pdf)と仮想NGSデータ(kadai_20160720.fasta)を追加しました。 提出期限は8月20日です。
- 予習事項(必須)
- インストール | についての推奨手順
(Windows2018.03.12版とMacintosh2015.04.03版)に従って
フリーソフトRと必要なパッケージをインストール。本講義用にカスタマイズしてはいない
(そんなものを作る時間があったら、講義資料の充実に費やします)ので利用しないパッケージもありますが、
自己責任で不必要だと思われるパッケージを入れないのは自由です。
- 下記パッケージを利用予定です(2016年07月12日現在)。コピペしてロードできるかどうかを確認しておきましょう。
- 基本的な利用法(Windows2015.04.03版と
Macintosh2015.04.03版)をやっておく。
- 平成27年度講習会の「データ解析環境R(7月29-30日分)」の自習。
- 2015年7月29日分(R基礎)の講義資料PDF (2015.07.27版;約7MB)
- 2015年7月30日分(Bioconductorの利用法)の講義資料PDF (2015.07.31版;約7MB)
- 2日分共通のhoge.zip(約75MB)
- チェックリスト
- インストール | についてをよく読み、ここに書いてある手順に従って最低限2015年4月4日以降にインストールを行った。
- 2016年5月11日以降に推奨手順通りに行うとR ver. 3.3.0以上がインストールされ(2016年7月9日現在はver. 3.3.1)、講義資料と同じ結果が得られる可能性が高いと知っている。
- インストール | についてで書いてある内容はBio-Linux8(ゲストOS)とは無関係であり、 WindowsやMacintosh(つまりホストOS)上で行う作業である。
- ファイルの拡張子(.txtや.docxなど)はちゃんと表示されている。
- Rの起動と終了ができる。終了時に表示されるメッセージにうろたえない。
- R本体だけでなくRパッケージ群のインストールもちゃんと行った。
- Rパッケージ群のインストール確認も行い、エラーが出ないことを確認した。
- library関数を用いたRパッケージのロード中に、別のパッケージがないことに起因するエラーメッセージが出ることもあるが、 必要なパッケージを個別にインストールするやり方を知っている。
- 基本的な利用法をよく読み、予習を行った。
- 作業ディレクトリの変更ができる。getwd()でちゃんと変更の確認ができる。
- ファイルのダウンロード時に、拡張子が勝手に変わることがあるので注意する(講義資料と同じ拡張子に自力で修正できる)。
- 慣れないうちは、getwd()とlist.files()を打ち込むことで、作業ディレクトリと入力ファイルの存在確認を行う。ファイルの拡張子もちゃんと確認する。
- エラーに遭遇した際、「ありがちなミス1-4」に当てはまっていないかどうか自分で確認し、対応できる。
- 第1部の心構え(掟)
- 予習事項(推奨)
- 2016年07月20日分(ゲノム解析、塩基配列解析)の講義資料PDF(2016.07.12版; 約7MB)
- 2016年07月20日分(ゲノム解析、塩基配列解析)のhoge.zip(2016.05.13版; 約2MB)
- 2016年07月21日分(トランスクリプトーム解析1)の講義資料PDF(2016.07.12版; 約7MB)
- 2016年07月21日分(トランスクリプトーム解析1)のhoge.zip(2016.05.23版; 約3MB)
- 2016年07月22日分(トランスクリプトーム解析2)の講義資料PDF(2016.07.13版; 約7MB)
- 2016年07月22日分(トランスクリプトーム解析2)のhoge.zip(2016.05.23版; 約1MB)
- その他(注意事項)
- Rは、バージョンやOSの違いによって解析実行時の挙動や結果が多少異なります。 本講義資料は2016年5月3日リリースのR ver. 3.3.0で作成し、動作確認を行っております。
- できるだけ講習会資料と同じ結果を得たい、持ち込みPCのヒトはこのバージョンのRをインストールしておきましょう。
- HDD容量に余裕がある方は、複数のバージョンをインストールしておくと、様々な挙動を体感できるかもしれません。
- アグリバイオ(主催機関)の貸与PC (Windows 7)はR ver. 3.3.0ですので、動作確認用に貸与希望宣言しておくのも一手です。
- 講師(門田)は、Windowsユーザです。Macintosh環境での動作確認は行っておりません。
- 例年Macintoshユーザの一部は、(第1部に限らず)なんらかの不具合に遭遇しますので、Macintoshユーザは覚悟しておいてください。
- 講義補助員(TA)がMacintoshユーザとは限りませんので、予めご了承ください。
- Macユーザのヒトは、アグリバイオPCを借り、7月19日の時間を使ってWindows環境に慣れておき、自身のMacと貸与されたWindows PCの両方でハンズオンを行うのも一手です。
- 主に講義資料の上部に「教科書pXX」などと書いていますが、これは平成28年度参考図書の
「トランスクリプトーム解析」のことを指します。
必要に応じて指示されたページ周辺を事前に読んでおくか、後で復習するなりしてください。
- アグリバイオ大学院講義科目「農学生命情報科学特論I (特論I)」受講者用
講習会期間中、ノートPC (Windows 7)の貸与を希望する方も、きっちり目を通しておきましょう。7月19日の自習日に貸与PCに慣れておくことを推奨。
library(Biostrings) #2016.07.20に利用 library(BSgenome.Hsapiens.NCBI.GRCh38) #2016.07.20に利用 library(MBCluster.Seq) #2016.07.22に利用 library(TCC) #2016.07.21と2016.07.22に利用
20160720_22_okite_20160618.pdf(2016.06.18版;約1MB)
参加予定の平成28年度講習会資料に目を通しておきましょう。随時差し替えます。 持込PCのヒトは、hoge.zipをダウンロード・解凍しておき、講義資料の1ページ目で示した状態にしてスムーズに講義がスタートできるようにしておきましょう。 貸与PCのヒトは、デスクトップ上にある「NGS28というフォルダ内にある各日のhogeフォルダ」をデスクトップにコピーしておき、 講義資料の1ページ目で示した状態にしてスムーズに講義がスタートできるようにしておきましょう。
特論Iの課題(kadai_tokuronI.pdf)と仮想NGSデータ(kadai_20160720.fasta)を2016年7月14日に公開しました。
提出期限は8月20日です。
第1部 | 統計解析 | ゲノム解析、塩基配列解析(2016.07.20)
当日は講義資料のスライド25あたりからやる予定です。スライド24あたりまでは自習。 スライド57のBSgenome.Hsapiens.NCBI.GRCh38でパッケージのインストールができていなかったときは、 PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB)の項目7-2を参考にしてパッケージのインストールを試みてください。
- 講義資料PDF(2016.07.12版; 約7MB)
- hoge.zip(2016.05.13版; 約2MB)
- NGSデータ解析戦略 (スライド6)
- DDBJ Pipeline:Nagasaki et al., DNA Res., 2013
- Illumina BaseSpace
- Galaxy:Goecks et al., Genome Biol., 2010
- NGSデータ (スライド9)
- Lactobacillus hokkaidonensis LOOC260(T):Tanizawa et al., BMC Genomics, 2015
- de novoアセンブリ (スライド24)
- 日本乳酸菌学会誌のNGS連載第6回ゲノムアセンブリ (スライド27)
- DDBJ PipelineでPlatanus (スライド28)
- Platanus ver. 1.2.2を実行したzip圧縮ファイル(platanusResult.zip; 約2.2MB)
- k-mer解析(k=1) (スライド33)
- 一気に結果を得る (スライド42)
- 配列数 (スライド49)
- k-mer解析(k=2) (スライド53)
- k-mer解析(k=2) (スライド56)
- k-mer解析(k=2) (スライド57)
- 作図(box plot):基本形 (スライド59)
- 作図(box plot):色づけ (スライド64)
- 作図(box plot):発展形 (スライド68)
- ランダムデータ生成 (スライド72)
- k-mer解析(k=2) (スライド90)
- k-mer解析(k=2) (スライド93)
- k-mer解析(k=3) (スライド95)
- ランダムデータ生成 (スライド99)
- k-mer解析(k=3) (スライド102)
- k-mer解析(k=10) (スライド107)
- ランダムデータ生成 (スライド109)
- k-mer解析(k=10) (スライド112)
- k=6, 8, 10, 12 (スライド120)
- 作図(k=6, 8, 10) (スライド124)
- 塩基置換 (スライド128)
- 縦軸の範囲を揃える (スライド138)
- 最終確認 (スライド145)
塩基ごとの出現頻度解析は、k-mer解析のk=1の場合に相当します。 「イントロ | 一般 | k-mer解析 | k=1(塩基ごとの出現頻度解析) | Biostrings」の例題7は、以下と同じ。 入力ファイルはout_gapClosed.fa (約2.4MB)
in_f <- "out_gapClosed.fa" #入力ファイル名を指定してin_fに格納 out_f <- "hoge7.txt" #出力ファイル名を指定してout_fに格納 param_base <- c("A", "C", "G", "T", "N")#出力させたい塩基を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番 hoge <- alphabetFrequency(fasta) #A,C,G,T,..の数を各配列ごとにカウントした結果をhogeに格納 obj <- is.element(colnames(hoge), param_base)#条件を満たすかどうかを判定した結果をobjに格納 #out <- colSums(hoge[, obj]) #列ごとの総和をoutに格納 out <- apply(as.matrix(hoge[, obj]), 2, sum)#列ごとの総和をoutに格納 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#tmpの中身を指定したファイル名で保存
実際の利用時は、rcode1.txtのような無駄なコメントを除いてスリムにした一連のスクリプトを作成しておき、一気にコピペする。 ファイルの中身は以下と同じ。
param_base <- c("A", "C", "G", "T", "N")#出力させたい塩基を指定
library(Biostrings) #パッケージの読み込み
################
### Step 1
################
in_f <- "out_contig.fa" #入力ファイル名を指定してin_fに格納
out_f <- "result_step1.txt" #出力ファイル名を指定してout_fに格納
fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み
hoge <- alphabetFrequency(fasta) #A,C,G,T,..の数を各配列ごとにカウントした結果をhogeに格納
obj <- is.element(colnames(hoge), param_base)#条件を満たすかどうかを判定した結果をobjに格納
out <- apply(as.matrix(hoge[, obj]), 2, sum)#列ごとの総和をoutに格納
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存
################
### Step 2
################
in_f <- "out_scaffold.fa" #入力ファイル名を指定してin_fに格納
out_f <- "result_step2.txt" #出力ファイル名を指定してout_fに格納
fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み
hoge <- alphabetFrequency(fasta) #A,C,G,T,..の数を各配列ごとにカウントした結果をhogeに格納
obj <- is.element(colnames(hoge), param_base)#条件を満たすかどうかを判定した結果をobjに格納
out <- apply(as.matrix(hoge[, obj]), 2, sum)#列ごとの総和をoutに格納
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存
################
### Step 3
################
in_f <- "out_gapClosed.fa" #入力ファイル名を指定してin_fに格納
out_f <- "result_step3.txt" #出力ファイル名を指定してout_fに格納
fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み
hoge <- alphabetFrequency(fasta) #A,C,G,T,..の数を各配列ごとにカウントした結果をhogeに格納
obj <- is.element(colnames(hoge), param_base)#条件を満たすかどうかを判定した結果をobjに格納
out <- apply(as.matrix(hoge[, obj]), 2, sum)#列ごとの総和をoutに格納
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存
rcode2.txtは、配列数をカウントする必要最小限のコード。 ファイルの中身は以下と同じ。
library(Biostrings) #パッケージの読み込み ### Step 1 ### in_f <- "out_contig.fa" #入力ファイル名を指定してin_fに格納 fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み length(fasta) #配列数を表示 ### Step 2 ### in_f <- "out_scaffold.fa" #入力ファイル名を指定してin_fに格納 fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み length(fasta) #配列数を表示 ### Step 2 ### in_f <- "out_gapClosed.fa" #入力ファイル名を指定してin_fに格納 fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み length(fasta) #配列数を表示
2連続塩基の出現頻度解析は、k-mer解析のk=2の場合に相当します。 「イントロ | 一般 | k-mer解析 | k=2(2連続塩基の出現頻度解析) | Biostrings」の例題1(出現頻度)は、以下と同じ。 入力ファイルはhoge4.fa
in_f <- "hoge4.fa" #入力ファイル名を指定してin_fに格納 out_f <- "hoge1.txt" #出力ファイル名を指定してout_fに格納 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み fasta #確認してるだけです #本番 out <- dinucleotideFrequency(fasta) #連続塩基の出現頻度情報をoutに格納 #ファイルに保存 tmp <- cbind(names(fasta), out) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=2(2連続塩基の出現頻度解析) | Biostrings」の例題2(出現確率)は、以下と同じ。 入力ファイルはhoge4.fa
in_f <- "hoge4.fa" #入力ファイル名を指定してin_fに格納 out_f <- "hoge2.txt" #出力ファイル名を指定してout_fに格納 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み fasta #確認してるだけです #本番 out <- dinucleotideFrequency(fasta, as.prob=T)#連続塩基の出現確率情報をoutに格納 #ファイルに保存 tmp <- cbind(names(fasta), out) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=2(2連続塩基の出現頻度解析) | Biostrings」の例題7(出現確率)は、以下と同じ。
out_f <- "hoge7.txt" #出力ファイル名を指定してout_fに格納 param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"#パッケージ名を指定(BSgenome系のゲノムパッケージ) #必要なパッケージをロード library(Biostrings) #パッケージの読み込み library(param_bsgenome, character.only=T)#指定したパッケージの読み込み #前処理(指定したパッケージ中のオブジェクト名をgenomeに統一) tmp <- ls(paste("package", param_bsgenome, sep=":"))#指定したパッケージで利用可能なオブジェクト名を取得した結果をtmpに格納 genome <- eval(parse(text=tmp)) #文字列tmpをRオブジェクトとしてgenomeに格納(パッケージ中にオブジェクトが一つしかないという前提です) fasta <- getSeq(genome) #ゲノム塩基配列情報を抽出した結果をfastaに格納 names(fasta) <- seqnames(genome) #description情報を追加している fasta #確認してるだけです #本番 out <- dinucleotideFrequency(fasta, as.prob=T)#連続塩基の出現確率情報をoutに格納 #ファイルに保存 tmp <- cbind(names(fasta), out) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=2(2連続塩基の出現頻度解析) | Biostrings」の例題10(出現確率とbox plotのPNGファイル)は、以下と同じ。
out_f1 <- "hoge10.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge10.png" #出力ファイル名を指定してout_f2に格納 param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"#パッケージ名を指定(BSgenome系のゲノムパッケージ) param_fig <- c(700, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(Biostrings) #パッケージの読み込み library(param_bsgenome, character.only=T)#指定したパッケージの読み込み #前処理(指定したパッケージ中のオブジェクト名をgenomeに統一) tmp <- ls(paste("package", param_bsgenome, sep=":"))#指定したパッケージで利用可能なオブジェクト名を取得した結果をtmpに格納 genome <- eval(parse(text=tmp)) #文字列tmpをRオブジェクトとしてgenomeに格納(パッケージ中にオブジェクトが一つしかないという前提です) fasta <- getSeq(genome) #ゲノム塩基配列情報を抽出した結果をfastaに格納 names(fasta) <- seqnames(genome) #description情報を追加している fasta #確認してるだけです #本番 out <- dinucleotideFrequency(fasta, as.prob=T)#連続塩基の出現確率情報をoutに格納 #ファイルに保存(テキストファイル) tmp <- cbind(names(fasta), out) #保存したい情報をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(pngファイル) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 boxplot(out, ylab="Probability") #描画 grid(col="gray", lty="dotted") #指定したパラメータでグリッドを表示 dev.off() #おまじない
「イントロ | 一般 | k-mer解析 | k=2(2連続塩基の出現頻度解析) | Biostrings」の例題11(色分けして作図)は、以下と同じ。 連続塩基の種類ごとの期待値とボックスプロット(box plot)上での色情報を含むファイル (human_2mer.txt)を入力として利用し、色情報のみを取り出して利用しています。
in_f <- "human_2mer.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge11.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge11.png" #出力ファイル名を指定してout_f2に格納 param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"#パッケージ名を指定(BSgenome系のゲノムパッケージ) param_fig <- c(700, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(Biostrings) #パッケージの読み込み library(param_bsgenome, character.only=T)#指定したパッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(指定したパッケージ中のオブジェクト名をgenomeに統一) tmp <- ls(paste("package", param_bsgenome, sep=":"))#指定したパッケージで利用可能なオブジェクト名を取得した結果をtmpに格納 genome <- eval(parse(text=tmp)) #文字列tmpをRオブジェクトとしてgenomeに格納(パッケージ中にオブジェクトが一つしかないという前提です) fasta <- getSeq(genome) #ゲノム塩基配列情報を抽出した結果をfastaに格納 names(fasta) <- seqnames(genome) #description情報を追加している fasta #確認してるだけです #本番 out <- dinucleotideFrequency(fasta, as.prob=T)#連続塩基の出現確率情報をoutに格納 #ファイルに保存(テキストファイル) tmp <- cbind(names(fasta), out) #保存したい情報をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(pngファイル) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 boxplot(out, ylab="Probability", col=as.character(data$color))#描画 grid(col="gray", lty="dotted") #指定したパラメータでグリッドを表示 dev.off() #おまじない
「イントロ | 一般 | k-mer解析 | k=2(2連続塩基の出現頻度解析) | Biostrings」の例題12(期待値で割る)は、以下と同じ。 連続塩基の種類ごとの期待値とボックスプロット(box plot)上での色情報を含むファイル (human_2mer.txt)を入力として利用し、色情報のみを取り出して利用しています。 要なのは期待値からの差分であり、「プロットも期待値(expected)と同程度の観測値(observed)であればゼロ、 観測値のほうが大きければプラス、観測値のほうが小さければマイナス」といった具合で表現したほうがスマートです。 それゆえ、box plotの縦軸をlog(observed/expected)として表現しています。 CG以外の連続塩基は縦軸上でが0近辺に位置していることが分かります。
in_f <- "human_2mer.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge12.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge12.png" #出力ファイル名を指定してout_f2に格納 param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"#パッケージ名を指定(BSgenome系のゲノムパッケージ) param_fig <- c(700, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(Biostrings) #パッケージの読み込み library(param_bsgenome, character.only=T)#指定したパッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(指定したパッケージ中のオブジェクト名をgenomeに統一) tmp <- ls(paste("package", param_bsgenome, sep=":"))#指定したパッケージで利用可能なオブジェクト名を取得した結果をtmpに格納 genome <- eval(parse(text=tmp)) #文字列tmpをRオブジェクトとしてgenomeに格納(パッケージ中にオブジェクトが一つしかないという前提です) fasta <- getSeq(genome) #ゲノム塩基配列情報を抽出した結果をfastaに格納 names(fasta) <- seqnames(genome) #description情報を追加している fasta #確認してるだけです #本番 out <- dinucleotideFrequency(fasta, as.prob=T)#連続塩基の出現確率情報をoutに格納 #後処理(log(observed/expected)に変換) logratio <- log2(out/data$expected) #log(observed/expected)の計算結果をlogratioに格納 #ファイルに保存(テキストファイル) tmp <- cbind(names(fasta), logratio) #保存したい情報をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(pngファイル) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 boxplot(logratio, ylab="log2(observed/expected)",#描画 col=as.character(data$color)) #描画 grid(col="gray", lty="dotted") #指定したパラメータでグリッドを表示 dev.off() #おまじない
「サンプルデータ」の例題32は、以下と同じ。 出力ファイルは、sample32_ref.fastaとsample32_ngs.fastaです。
out_f1 <- "sample32_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample32_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 50 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 20 #リード長を指定 param_num_ngs <- 10 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=n(n連続塩基の出現頻度解析) | Biostrings」の例題7は、以下と同じ。
in_f <- "sample32_ngs.fasta" #入力ファイル名を指定してin_fに格納 out_f <- "hoge7.txt" #出力ファイル名を指定してout_fに格納 param_kmer <- 2 #k-merのkの値を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番 out <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をoutに格納 #ファイルに保存 tmp <- cbind(names(fasta), out) #最初の列にID情報、そのあとに出現頻度情報のoutを結合したtmpを作成 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=n(n連続塩基の出現頻度解析) | Biostrings」の例題8は、以下と同じ。
in_f <- "sample32_ngs.fasta" #入力ファイル名を指定してin_fに格納 out_f <- "hoge8.txt" #出力ファイル名を指定してout_fに格納 param_kmer <- 2 #k-merのkの値を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=n(n連続塩基の出現頻度解析) | Biostrings」の例題9は、以下と同じ。
in_f <- "sample32_ngs.fasta" #入力ファイル名を指定してin_fに格納 out_f <- "hoge9.txt" #出力ファイル名を指定してout_fに格納 param_kmer <- 3 #k-merのkの値を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存
「サンプルデータ」の例題33は、以下と同じ。 出力ファイルは、sample33_ref.fastaとsample33_ngs.fastaです。
out_f1 <- "sample33_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample33_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 1000 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 20 #リード長を指定 param_num_ngs <- 200 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=n(n連続塩基の出現頻度解析) | Biostrings」の例題10は、以下と同じ。
in_f <- "sample33_ngs.fasta" #入力ファイル名を指定してin_fに格納 out_f <- "hoge10.txt" #出力ファイル名を指定してout_fに格納 param_kmer <- 3 #k-merのkの値を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=n(n連続塩基の出現頻度解析) | Biostrings」の例題11は、以下と同じ。
in_f <- "sample33_ngs.fasta" #入力ファイル名を指定してin_fに格納 out_f <- "hoge11.txt" #出力ファイル名を指定してout_fに格納 param_kmer <- 10 #k-merのkの値を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存 length(out) #4^param_kmerの値を表示 sum(out > 0) #1回以上出現したk-merの種類数を表示
「サンプルデータ」の例題34は、以下と同じ。 出力ファイルは、sample34_ref.fastaとsample34_ngs.fastaです。
out_f1 <- "sample34_ref.fasta" #出力ファイル名を指定してout_f1に格納 out_f2 <- "sample34_ngs.fasta" #出力ファイル名を指定してout_f2に格納 param_len_ref <- 1000 #リファレンス配列の長さを指定 narabi <- c("A","C","G","T") #以下の数値指定時にACGTの並びを間違えないようにするために表示(内部的にも使用してます) param_composition <- c(22, 28, 28, 22) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param_len_ngs <- 20 #リード長を指定 param_num_ngs <- 500 #リード数を指定 param_desc <- "kkk" #FASTA形式ファイルのdescription行に記述する内容 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #本番(リファレンス配列生成) set.seed(1010) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param_composition)#narabi中の塩基がparam_compositionで指定した数だけ存在する文字列ベクトルACGTsetを作成 reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")#ACGTsetからparam_len_ref回分だけ復元抽出して得られた塩基配列をreferenceに格納 reference <- DNAStringSet(reference) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をreferenceに格納 names(reference) <- param_desc #description行に相当する記述を追加している reference #確認してるだけです #本番(シミュレーションデータ生成) s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)#部分塩基配列抽出時のstart position情報として用いる乱数ベクトルをs_posiに格納 s_posi #確認してるだけです hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:length(s_posi)){ #length(s_posi)回だけループを回す hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))#subseq関数を用いてs_posi[i]からparam_len_ngsで与えた配列長に対応する部分配列をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 #後処理(description部分の作成) description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")#param_descやs_posiなどを"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している fasta #確認してるだけです #ファイルに保存(仮想リファレンス配列と仮想NGS配列) writeXStringSet(reference, file=out_f1, format="fasta", width=50)#referenceの中身を指定したファイル名で保存 writeXStringSet(fasta, file=out_f2, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
「イントロ | 一般 | k-mer解析 | k=n(n連続塩基の出現頻度解析) | Biostrings」の例題12は、以下と同じ。
in_f <- "sample34_ngs.fasta" #入力ファイル名を指定してin_fに格納 out_f <- "hoge12.txt" #出力ファイル名を指定してout_fに格納 param_kmer <- 10 #k-merのkの値を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 #ファイルに保存 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)#outの中身を指定したファイル名で保存 length(out) #4^param_kmerの値を表示 sum(out > 0) #1回以上出現したk-merの種類数を表示
必要最小限のコードにして、k値の違い(k=6, 8, 10, and 12)による影響の全体像を把握。
in_f <- "sample34_ngs.fasta" #入力ファイル名を指定してin_fに格納 library(Biostrings) #パッケージの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み param_kmer <- 6 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 median(kmer) #出現回数の中央値(median)を表示 param_kmer <- 8 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 median(kmer) #出現回数の中央値(median)を表示 param_kmer <- 10 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 median(kmer) #出現回数の中央値(median)を表示 param_kmer <- 12 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 median(kmer) #出現回数の中央値(median)を表示
上記のk=6, 8, 10の結果より、横軸の最大値は39であることがわかっているが、ヒストグラム全体の変遷が把握しづらいので 1から20の範囲を指定している。このあたりは結果を眺めながら自分の好みに合わせて微調整する。
in_f <- "sample34_ngs.fasta" #入力ファイル名を指定してin_fに格納 param_fig <- c(450, 210) #ヒストグラム描画時の横幅と縦幅を指定(単位はピクセル) param_range <- c(1, 20) #表示したい横軸の範囲を指定 library(Biostrings) #パッケージの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み out_f <- "hoge1.png" #出力ファイル名を指定してout_fに格納 param_kmer <- 6 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_range, main="") #ヒストグラムを描画 dev.off() #おまじない out_f <- "hoge2.png" #出力ファイル名を指定してout_fに格納 param_kmer <- 8 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_range, main="") #ヒストグラムを描画 dev.off() #おまじない out_f <- "hoge3.png" #出力ファイル名を指定してout_fに格納 param_kmer <- 10 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_range, main="") #ヒストグラムを描画 dev.off() #おまじない
GならC、AならTのような塩基置換を行うenkichikan関数(孫 建強氏作成)を利用して任意の位置の塩基置換を行う。 「書籍 | トランスクリプトーム解析 | 2.3.4 マッピング(準備)」のp81と同じ関数です。
in_f <- "sample34_ngs.fasta" #入力ファイル名を指定してin_fに格納 param_fig <- c(450, 210) #ヒストグラム描画時の横幅と縦幅を指定(単位はピクセル) param_range <- c(1, 20) #表示したい横軸の範囲を指定 library(Biostrings) #パッケージの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み fasta #確認してるだけです #塩基置換関数の作成 enkichikan <- function(fa, p) { #関数名や引数の作成 t <- substring(fa, p, p) #置換したい位置の塩基を取りだす t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成 substring(fa, p, p) <- t_c #置換 return(fa) #置換後のデータを返す } #enkichikan関数の動作確認 as.character(fasta[500]) #500番目(最後)のリードを文字列として表示 enkichikan(as.character(fasta[500]), 18)#18番目の塩基を置換 #塩基置換本番 fasta_org <- fasta #塩基置換前のfastaをfasta_orgに格納 hoge <- enkichikan(as.character(fasta), 18)#18番目の塩基を置換 fasta <- DNAStringSet(hoge) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をfastaに格納 names(fasta) <- names(fasta_org) #fasta_orgのdescription情報をコピー fasta #確認してるだけです #k-mer出現頻度分布 out_f <- "hoge2_18.png" #出力ファイル名を指定してout_fに格納 param_kmer <- 8 #k-merのkの値を指定 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_range, main="") #ヒストグラムを描画 dev.off() #おまじない
ついでに、x軸の範囲指定のところの名前をparam_rangeからparam_xrangeに変更しています。
in_f <- "sample34_ngs.fasta" #入力ファイル名を指定してin_fに格納 param_fig <- c(450, 210) #ヒストグラム描画時の横幅と縦幅を指定(単位はピクセル) param_xrange <- c(1, 20) #表示したい横軸の範囲を指定 param_yrange <- c(0, 250) #表示したい縦軸の範囲を指定 param_kmer <- 8 #k-merのkの値を指定 library(Biostrings) #パッケージの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み fasta #確認してるだけです #塩基置換関数の作成 enkichikan <- function(fa, p) { #関数名や引数の作成 t <- substring(fa, p, p) #置換したい位置の塩基を取りだす t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成 substring(fa, p, p) <- t_c #置換 return(fa) #置換後のデータを返す } #塩基置換本番 fasta_org <- fasta #塩基置換前のfastaをfasta_orgに格納 hoge <- enkichikan(as.character(fasta), 18)#18番目の塩基を置換 fasta <- DNAStringSet(hoge) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をfastaに格納 names(fasta) <- names(fasta_org) #fasta_orgのdescription情報をコピー fasta #確認してるだけです #k-mer出現頻度分布(塩基置換後) out_f <- "hoge2_18_250.png" #出力ファイル名を指定してout_fに格納 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_xrange, ylim=param_yrange, main="")#ヒストグラムを描画 dev.off() #おまじない #k-mer出現頻度分布(塩基置換前) fasta <- fasta_org #塩基置換前のfasta_orgをfastaに格納 out_f <- "hoge2_250.png" #出力ファイル名を指定してout_fに格納 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_xrange, ylim=param_yrange, main="")#ヒストグラムを描画 dev.off() #おまじない
500リードのうち、451:500番目のリードについてのみ13番目の塩基を置換。
in_f <- "sample34_ngs.fasta" #入力ファイル名を指定してin_fに格納 param_fig <- c(450, 210) #ヒストグラム描画時の横幅と縦幅を指定(単位はピクセル) param_xrange <- c(1, 20) #表示したい横軸の範囲を指定 param_yrange <- c(0, 350) #表示したい縦軸の範囲を指定 param_kmer <- 8 #k-merのkの値を指定 library(Biostrings) #パッケージの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み fasta #確認してるだけです fasta_first450 <- fasta[1:450] #塩基置換を行わない最初の450リード分を抽出してfasta_first450に格納 fasta_first450 #確認してるだけです #塩基置換関数の作成 enkichikan <- function(fa, p) { #関数名や引数の作成 t <- substring(fa, p, p) #置換したい位置の塩基を取りだす t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成 substring(fa, p, p) <- t_c #置換 return(fa) #置換後のデータを返す } #塩基置換本番(451:500のみ) fasta_org <- fasta #塩基置換前のfastaをfasta_orgに格納 hoge <- enkichikan(as.character(fasta[451:500]), 13)#13番目の塩基を置換 fasta <- DNAStringSet(hoge) #DNA塩基配列だと認識させるDNAStringSet関数を適用した結果をfastaに格納 names(fasta) <- names(fasta_org[451:500])#fasta_orgのdescription情報をコピー fasta_last50 <- fasta #fasta_last50として取り扱う fasta_last50 #確認してるだけです #k-mer出現頻度分布(original) fasta <- fasta_org #fasta_orgをfastaに格納 out_f <- "hoge2_original.png" #出力ファイル名を指定してout_fに格納 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_xrange, ylim=param_yrange, main="")#ヒストグラムを描画 dev.off() #おまじない kmer_org <- kmer #kmerをkmer_orgに格納 #k-mer出現頻度分布(最初の450リードのみ;塩基置換なし) fasta <- fasta_first450 #fasta_first450をfastaに格納 out_f <- "hoge2_first450.png" #出力ファイル名を指定してout_fに格納 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_xrange, ylim=param_yrange, main="")#ヒストグラムを描画 dev.off() #おまじない kmer_first450 <- kmer #kmerをkmer_first450に格納 #k-mer出現頻度分布(最後の50リードのみ;塩基置換あり) fasta <- fasta_last50 #fasta_last50をfastaに格納 out_f <- "hoge2_last50.png" #出力ファイル名を指定してout_fに格納 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_xrange, ylim=param_yrange, main="")#ヒストグラムを描画 dev.off() #おまじない kmer_last50 <- kmer #kmerをkmer_last50に格納 #k-mer出現頻度分布(merged) fasta <- c(fasta_first450, fasta_last50)#マージしたものをfastaに格納 out_f <- "hoge2_merged.png" #出力ファイル名を指定してout_fに格納 hoge <- oligonucleotideFrequency(fasta, width=param_kmer)#k連続塩基の出現頻度情報をhogeに格納 out <- colSums(hoge) #列ごとの総和をoutに格納 kmer <- out[out > 0] #1回以上出現したk-merのみをkmerに格納 length(kmer) #1回以上出現したk-merの種類数を表示 table(kmer) #(k-merの種類は問わずに)k-merの出現回数分布を表示 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #下、左、上、右の順で余白(行)を指定 hist(kmer, breaks=max(kmer), ylab="Frequency",#ヒストグラムを描画 xlab=paste("Number of occurences at k=", param_kmer, sep=""),#ヒストグラムを描画 xlim=param_xrange, ylim=param_yrange, main="")#ヒストグラムを描画 dev.off() #おまじない kmer_merged <- kmer #kmerをkmer_mergedに格納
第1部 | 統計解析 | トランスクリプトーム解析1(2016.07.21)
平成27年度講習会(2015.07.29のR:基礎のスライド46-92あたり、および2015.08.05のRNA-seq:統計解析)のをベースとしています。
- 講義資料PDF(2016.07.12版; 約7MB)
- hoge.zip(2016.05.23版; 約3MB)
- 数値行列 (スライド5)
- データの正規化 (スライド8)
- RPM正規化 (スライド14)
- RPKM正規化 (スライド18)
- クラスタリング(サンプル間) (スライド21)
- サブセット抽出と整形 (スライド25)
- クラスタリング(サンプル間) (スライド27)
- クラスタリングとDEG数の関係 (スライド32)
- Tips: cex.lab (スライド33)
- Tips: mar (スライド34)
- ReCount:Frazee et al., BMC Bioinformatics, 2011 (スライド36)
- bodymapデータ解析 (スライド39)
- TCCで実行すると... (スライド41)
- ReCountのヒトデータは (スライド42)
- giladデータ解析 (スライド45)
- giladデータ解析 (スライド46)
- ReCountのヒトデータは (スライド53)
- bodymap + gilad (スライド55)
- HS vs. RM (スライド64)
- DEG数の見積もり (スライド71)
- DEG検出結果1位 (スライド77)
- DEG検出結果2位 (スライド78)
- DEG検出結果3位 (スライド79)
- DEG検出結果2,489位 (スライド80)
- 分布やモデル (スライド82)
- HS vs. PT (スライド85)
- サンプル間類似度RM (スライド92)
- サンプル間類似度PT (スライド93)
- HS1 vs. HS2 (スライド97)
- PT1 vs. PT2 (スライド98)
- RM1 vs. RM2 (スライド99)
- RM1 vs. RM3 (スライド100)
- 統計的手法とは (スライド103)
- 結果の比較(倍率変化とFDR) (スライド105)
- giladデータでDEG同定 (スライド109)
- クラスタリング(maqc) (スライド116)
- クラスタリング(blekhman) (スライド118)
- maqcデータでDEG同定 (スライド120)
- DEG数の見積もり(maqcデータ) (スライド124)
- mergeの基本形 (スライド127)
- maqc (pooled) (スライド133)
- maqc (pooled)でDEG同定 (スライド135)
- 反復あり or なし(TCC) (スライド143)
- 反復あり or なし(edgeR) (スライド145)
- 反復あり or なし(DESeq2) (スライド146)
- 反復あり or なし(DESeq2) (スライド148)
「サンプルデータ」の例題41は、以下と同じ。 Blekhman et al., Genome Res., 2010の Supplementary Table1で提供されているエクセルファイル(http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls; 約4.3MB) からカウントデータのみ抽出し、きれいに整形しなおしたものがここでの出力ファイルになります。 20,689 genes×36 samplesのカウントデータ(sample_blekhman_36.txt)です。
#in_f <- "http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls"#入力ファイル名を指定してin_fに格納 in_f <- "suppTable1.xls" #入力ファイル名を指定してin_fに格納 out_f <- "sample_blekhman_36.txt" #出力ファイル名を指定してout_fに格納 #入力ファイルの読み込み hoge <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(hoge) #行数と列数を表示 #サブセットの取得 data <- cbind( #必要な列名の情報を取得したい列の順番で結合した結果をdataに格納 hoge$R1L4.HSF1, hoge$R4L2.HSF1, hoge$R2L7.HSF2, hoge$R3L2.HSF2, hoge$R8L1.HSF3, hoge$R8L2.HSF3, hoge$R1L1.HSM1, hoge$R5L2.HSM1, hoge$R2L3.HSM2, hoge$R4L8.HSM2, hoge$R3L6.HSM3, hoge$R4L1.HSM3, hoge$R1L2.PTF1, hoge$R4L4.PTF1, hoge$R2L4.PTF2, hoge$R6L6.PTF2, hoge$R3L7.PTF3, hoge$R5L3.PTF3, hoge$R1L6.PTM1, hoge$R3L3.PTM1, hoge$R2L8.PTM2, hoge$R4L6.PTM2, hoge$R6L2.PTM3, hoge$R6L4.PTM3, hoge$R1L7.RMF1, hoge$R5L1.RMF1, hoge$R2L2.RMF2, hoge$R5L8.RMF2, hoge$R3L4.RMF3, hoge$R4L7.RMF3, hoge$R1L3.RMM1, hoge$R3L8.RMM1, hoge$R2L6.RMM2, hoge$R5L4.RMM2, hoge$R3L1.RMM3, hoge$R4L3.RMM3) colnames(data) <- c( #列名を付加 "R1L4.HSF1", "R4L2.HSF1", "R2L7.HSF2", "R3L2.HSF2", "R8L1.HSF3", "R8L2.HSF3", "R1L1.HSM1", "R5L2.HSM1", "R2L3.HSM2", "R4L8.HSM2", "R3L6.HSM3", "R4L1.HSM3", "R1L2.PTF1", "R4L4.PTF1", "R2L4.PTF2", "R6L6.PTF2", "R3L7.PTF3", "R5L3.PTF3", "R1L6.PTM1", "R3L3.PTM1", "R2L8.PTM2", "R4L6.PTM2", "R6L2.PTM3", "R6L4.PTM3", "R1L7.RMF1", "R5L1.RMF1", "R2L2.RMF2", "R5L8.RMF2", "R3L4.RMF3", "R4L7.RMF3", "R1L3.RMM1", "R3L8.RMM1", "R2L6.RMM2", "R5L4.RMM2", "R3L1.RMM3", "R4L3.RMM3") rownames(data)<- rownames(hoge) #行名を付加 dim(data) #行数と列数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
「書籍 | トランスクリプトーム解析 | 3.3.2 データの正規化(基礎編)」と説明内容は同じです。
2262/3 #スライド8:配列長の違いを考慮しないカウント比 (2262/6)/3 #スライド8:配列長の違いを考慮したカウント比 (2262/3000)/(3/500) #スライド8:配列長の違いを考慮したカウント比 #RPK補正 2262/3000 #スライド9:カウント数/配列長 3/500 #スライド9:カウント数/配列長 2262*(1000/3000) #スライド10:配列長が1000 bpだったときのカウント数 3*(1000/500) #スライド10:配列長が1000 bpだったときのカウント数 #RPM補正のイントロ colSums(data) #スライド13:列ごとの総リード数。スライド5コピペ時のdataオブジェクトを利用
「正規化 | 基礎 | RPM or CPM (総リード数補正)」をベースに作成。 入力は、20,689 genes×36 samplesのカウントデータ(sample_blekhman_36.txt)です。 教科書p134。
in_f <- "sample_blekhman_36.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge1.txt" #出力ファイル名を指定してout_fに格納 param1 <- 1000000 #補正後の総リード数を指定(RPMにしたい場合はここの数値はそのまま) #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み colSums(data) #総リード数を表示 #本番(正規化) nf <- param1/colSums(data) #正規化係数を計算した結果をnfに格納 data <- sweep(data, 2, nf, "*") #正規化係数を各列に掛けた結果をdataに格納 colSums(data) #総リード数を表示 #ファイルに保存 tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
行名が"ENSG00000000971"で、1列目のカウント値(2262)に対するRPKM値の計算例を示す。
#RPM補正部分 data["ENSG00000000971", 1] #dataから、行名が"ENSG00000000971"で、1列目の情報を表示 nf[1] #RPM正規化係数nfの1番目の要素を表示 2262 * nf[1] #"ENSG00000000971"の1列目のカウント情報(2262)に、対応するRPM正規化係数を掛けた結果を表示 2262 * (1000000/1665987) #"ENSG00000000971"の1列目のカウント情報(2262)に、対応するRPM正規化係数を掛けた結果を表示 #RPK補正追加分 1000/3000 #"ENSG00000000971"の配列長が3000だった場合のRPK正規化係数 data["ENSG00000000971", 1] * (1000/3000)#RPKM補正後の値を表示< 2262 * (1000000/1665987) * (1000/3000) #RPKM補正後の値を表示
「解析 | クラスタリング | サンプル間 | TCC(Sun_2013)」の例題7は以下と同じ。 入力は、20,689 genes×36 samplesのカウントデータ(sample_blekhman_36.txt)です。
in_f <- "sample_blekhman_36.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge7.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(700, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=1.2,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
「サンプルデータ」の例題42は以下と同じ。 入力は、Blekhman et al., Genome Res., 2010の Supplementary Table1で提供されているエクセルファイル(http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls; 約4.3MB) ですが、hogeフォルダ中にあります。例題41とは違って、technical replicatesの2列分のデータは足して1列分のデータとしています。 出力は、20,689 genes×18 samplesのカウントデータ(sample_blekhman_18.txt)です。
#in_f <- "http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls"#入力ファイル名を指定してin_fに格納 in_f <- "suppTable1.xls" #入力ファイル名を指定してin_fに格納 out_f <- "sample_blekhman_18.txt" #出力ファイル名を指定してout_fに格納 #入力ファイルの読み込み hoge <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(hoge) #行数と列数を表示 #サブセットの取得 data <- cbind( #必要な列名の情報を取得したい列の順番で結合した結果をdataに格納 hoge$R1L4.HSF1 + hoge$R4L2.HSF1, hoge$R2L7.HSF2 + hoge$R3L2.HSF2, hoge$R8L1.HSF3 + hoge$R8L2.HSF3, hoge$R1L1.HSM1 + hoge$R5L2.HSM1, hoge$R2L3.HSM2 + hoge$R4L8.HSM2, hoge$R3L6.HSM3 + hoge$R4L1.HSM3, hoge$R1L2.PTF1 + hoge$R4L4.PTF1, hoge$R2L4.PTF2 + hoge$R6L6.PTF2, hoge$R3L7.PTF3 + hoge$R5L3.PTF3, hoge$R1L6.PTM1 + hoge$R3L3.PTM1, hoge$R2L8.PTM2 + hoge$R4L6.PTM2, hoge$R6L2.PTM3 + hoge$R6L4.PTM3, hoge$R1L7.RMF1 + hoge$R5L1.RMF1, hoge$R2L2.RMF2 + hoge$R5L8.RMF2, hoge$R3L4.RMF3 + hoge$R4L7.RMF3, hoge$R1L3.RMM1 + hoge$R3L8.RMM1, hoge$R2L6.RMM2 + hoge$R5L4.RMM2, hoge$R3L1.RMM3 + hoge$R4L3.RMM3) colnames(data) <- c( #列名を付加 "HSF1", "HSF2", "HSF3", "HSM1", "HSM2", "HSM3", "PTF1", "PTF2", "PTF3", "PTM1", "PTM2", "PTM3", "RMF1", "RMF2", "RMF3", "RMM1", "RMM2", "RMM3") rownames(data)<- rownames(hoge) #行名を付加 dim(data) #行数と列数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(data), data) #保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存
「解析 | クラスタリング | サンプル間 | TCC(Sun_2013)」の例題8は以下と同じ。 入力は、20,689 genes×18 samplesのカウントデータ(sample_blekhman_18.txt)です。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge8.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(700, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=1.2,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
原著論文はTang et al., BMC Bioinformatics, 2015です。
cex.labは、この場合縦軸ラベル情報("Height")の文字の大きさをデフォルトの何倍にして表示するかを指定するオプション。 例えばデフォルトの2.0倍にしたいときは、cex.lab=2.0とすればよい。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(600, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=2.0,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
marの左部分の余白を4から5行分に変更することで、"Height"の文字が切れなくなる。 marはmargin (マージン)の意味です。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(600, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 5, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=2.0,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
マッピング済みの行列形式のカウントデータセットを多数提供しています。
getwd()
list.files(pattern="bodymap")
「書籍 | トランスクリプトーム解析 | 3.3.3 クラスタリング」の「p143-144の網掛け部分」は、以下と同じ。
in_f1 <- "bodymap_count_table.txt" in_f2 <- "bodymap_phenodata.txt" ### ファイルの読み込み ### data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") ### dataオブジェクトの列名を変更 ### colnames(data) <- phenotype$tissue.type ### フィルタリング(総カウント数が0でなく、ユニークな発現パターンをもつもののみ) ### obj <- rowSums(data) != 0 hoge <- unique(data[obj,]) ### クラスタリング ### data.dist <- as.dist(1 - cor(hoge, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out)
「書籍 | トランスクリプトーム解析 | 3.3.3 クラスタリング」の「p143-144の網掛け部分」を、 「解析 | クラスタリング | サンプル間 | TCC(Sun_2013)」 のように書くと以下のような感じになります。
in_f1 <- "bodymap_count_table.txt" #入力ファイル名を指定してin_f1に格納(カウントデータ) in_f2 <- "bodymap_phenodata.txt" #入力ファイル名を指定してin_f2に格納(サンプルラベル情報) out_f <- "hoge.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(600, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") phenotype #確認してるだけです colnames(data) <- phenotype$tissue.type#dataオブジェクトの列名を変更 #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=1.2,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
bodymapを含む、ReCountで提供されているヒトデータは同じ遺伝子数
dim(data) head(rownames(data))
getwd()
list.files(pattern="gilad")
giladデータのサンプル間クラスタリングを実行。phenotype情報は入力ファイルによってフォーマットが異なる点に注意。
in_f1 <- "gilad_count_table.txt" #入力ファイル名を指定してin_f1に格納(カウントデータ) in_f2 <- "gilad_phenodata.txt" #入力ファイル名を指定してin_f2に格納(サンプルラベル情報) out_f <- "hoge_gilad.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(600, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") phenotype #確認してるだけです colnames(data) <- paste(phenotype$gender, rownames(phenotype), sep="_")#dataオブジェクトの列名を変更 colnames(data) #確認してるだけです #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=1.2,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
giladを含む、ReCountで提供されているヒトデータは同じ遺伝子数
dim(data) head(rownames(data))
2つのデータセットを読み込んで、マージ(列方向で結合)して、サンプル間クラスタリングを実行。
out_f <- "hoge_merge.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(700, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み(bodymap) in_f1 <- "bodymap_count_table.txt" #入力ファイル名を指定してin_f1に格納(カウントデータ) in_f2 <- "bodymap_phenodata.txt" #入力ファイル名を指定してin_f2に格納(サンプルラベル情報) data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") phenotype #確認してるだけです colnames(data) <- phenotype$tissue.type#dataオブジェクトの列名を変更 colnames(data) #確認してるだけです dim(data) #行数と列数を表示 data_bodymap <- data #dataをdata_bodymapに格納 #入力ファイルの読み込み(gilad) in_f1 <- "gilad_count_table.txt" #入力ファイル名を指定してin_f1に格納(カウントデータ) in_f2 <- "gilad_phenodata.txt" #入力ファイル名を指定してin_f2に格納(サンプルラベル情報) data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") phenotype #確認してるだけです colnames(data) <- paste(phenotype$gender, rownames(phenotype), sep="_")#dataオブジェクトの列名を変更 colnames(data) #確認してるだけです dim(data) #行数と列数を表示 data_gilad <- data #dataをdata_giladに格納 #本番 data <- cbind(data_bodymap, data_gilad)#複数のデータセットを列方向で結合(cbind) colnames(data) #確認してるだけです dim(data) #行数と列数を表示 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=1.2,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題1は以下と同じ。ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)です。 1, 4, 13, 16 列目のデータのみ抽出しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge1.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge1.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10, 10),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
2489*(1 - 0.05) 3121*(1 - 0.10) 4049*(1 - 0.20) 4785*(1 - 0.30)
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題2は以下と同じ。ランキング1位の"ENSG00000208570"をハイライトさせるテクニックです。 1位のy軸の値(m.value)が11.29なので、y軸の範囲を[-10.0, 11.5]の範囲に変更する操作も行っています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge2.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge2.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(400, 310) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) param_geneid <- "ENSG00000208570" #ハイライトさせたいgene_IDを指定 param_col <- "magenta" #色を指定 param_cex <- 2 #点の大きさを指定(2なら通常の2倍、0.5なら通常の0.5倍) param_pch <- 20 #点の形状を指定(詳細はこちらとか) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 obj <- is.element(tcc$gene_id, param_geneid)#条件を満たすかどうかを判定した結果をobjに格納(param_geneidで指定した位置をTRUE) points(result$a.value[obj], result$m.value[obj],#条件を満たす点をハイライト pch=param_pch, cex=param_cex, col=param_col)#条件を満たす点をハイライト dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題3は以下と同じ。ランキング2位の"ENSG00000220191"をハイライトさせるテクニックです。 param_cexやparam_pchを変更しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge3.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge3.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(400, 310) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) param_geneid <- "ENSG00000220191" #ハイライトさせたいgene_IDを指定 param_col <- "magenta" #色を指定 param_cex <- 2.3 #点の大きさを指定(2なら通常の2倍、0.5なら通常の0.5倍) param_pch <- 15 #点の形状を指定(詳細はこちらとか) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 obj <- is.element(tcc$gene_id, param_geneid)#条件を満たすかどうかを判定した結果をobjに格納(param_geneidで指定した位置をTRUE) points(result$a.value[obj], result$m.value[obj],#条件を満たす点をハイライト pch=param_pch, cex=param_cex, col=param_col)#条件を満たす点をハイライト dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題4は以下と同じ。ランキング3位の"ENSG00000106366"をハイライトさせるテクニックです。 param_cexやparam_pchを変更しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge4.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge4.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(400, 310) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) param_geneid <- "ENSG00000106366" #ハイライトさせたいgene_IDを指定 param_col <- "magenta" #色を指定 param_cex <- 3.1 #点の大きさを指定(2なら通常の2倍、0.5なら通常の0.5倍) param_pch <- 18 #点の形状を指定(詳細はこちらとか) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 obj <- is.element(tcc$gene_id, param_geneid)#条件を満たすかどうかを判定した結果をobjに格納(param_geneidで指定した位置をTRUE) points(result$a.value[obj], result$m.value[obj],#条件を満たす点をハイライト pch=param_pch, cex=param_cex, col=param_col)#条件を満たす点をハイライト dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題5は以下と同じ。ランキング2,489位の"ENSG00000139445"をハイライトさせるテクニックです。 param_col, param_cex, param_pchなどを変更しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge5.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge5.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(400, 310) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) param_geneid <- "ENSG00000139445" #ハイライトさせたいgene_IDを指定 param_col <- "skyblue" #色を指定 param_cex <- 2.5 #点の大きさを指定(2なら通常の2倍、0.5なら通常の0.5倍) param_pch <- 17 #点の形状を指定(詳細はこちらとか) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 obj <- is.element(tcc$gene_id, param_geneid)#条件を満たすかどうかを判定した結果をobjに格納(param_geneidで指定した位置をTRUE) points(result$a.value[obj], result$m.value[obj],#条件を満たす点をハイライト pch=param_pch, cex=param_cex, col=param_col)#条件を満たす点をハイライト dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題6は以下と同じ。3種類のFDR閾値(0.0001, 0.05, 0.4)でのM-A plotを出力させています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge6_1.png" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge6_2.png" #出力ファイル名を指定してout_f2に格納 out_f3 <- "hoge6_3.png" #出力ファイル名を指定してout_f3に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- c(0.0001, 0.05, 0.4) #false discovery rate (FDR)閾値を指定 param_fig <- c(375, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger")#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR[1]) #FDR < param_FDR[1]を満たす遺伝子数を表示 sum(tcc$stat$q.value < param_FDR[2]) #FDR < param_FDR[2]を満たす遺伝子数を表示 sum(tcc$stat$q.value < param_FDR[3]) #FDR < param_FDR[3]を満たす遺伝子数を表示 #ファイルに保存(M-A plot) png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR[1], xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR[1], ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR[2], xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR[2], ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR[3], xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR[3], ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題7は以下と同じ。1, 4, 7, 10 列目のデータのみ抽出して、ヒト2サンプル(G1群:HSF1とHSM1) vs. チンパンジー2サンプル(G2群:PTF1とPTM1)の2群間比較を行っています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge7.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge7.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 7, 10) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
unique関数周辺については、教科書p143辺りで解説しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #行数と列数を表示 data <- unique(data) #ユニークな発現パターンのみにしている dim(data) #行数と列数を表示 cor(data$HSM1, data$HSF1, method="spearman")#HS群内(HSF1 vs. HSM1)のSpearman相関係数 cor(data$RMM1, data$RMF1, method="spearman")#RM群内(RMF1 vs. RMM1)のSpearman相関係数 cor(data$HSM1, data$RMM1, method="spearman")#HSM1 vs. RMM1のSpearman相関係数
unique関数周辺については、教科書p143辺りで解説しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #行数と列数を表示 data <- unique(data) #ユニークな発現パターンのみにしている dim(data) #行数と列数を表示 cor(data$HSM1, data$HSF1, method="spearman")#HS群内(HSF1 vs. HSM1)のSpearman相関係数 cor(data$PTM1, data$PTF1, method="spearman")#PT群内(PTF1 vs. PTM1)のSpearman相関係数 cor(data$HSM1, data$PTM1, method="spearman")#HSM1 vs. PTM1のSpearman相関係数
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題8は以下と同じ。1, 4, 2, 5 列目のデータのみ抽出して、ヒト2サンプル(G1群:HSF1とHSM1) vs. ヒト2サンプル(G2群:HSF2とHSM2)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge8.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge8.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 2, 5) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 obj <- as.logical(tcc$stat$q.value < param_FDR)#条件を満たすかどうかを判定した結果をobjに格納 points(result$a.value[obj], result$m.value[obj],#objがTRUEとなる点をハイライト pch=20, cex=1.5, col="magenta")#objがTRUEとなる点をハイライト legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題9は以下と同じ。7, 10, 8, 11列目のデータのみ抽出して、 チンパンジー2サンプル(G1群:PTF1とPTM1) vs. チンパンジー2サンプル(G2群:PTF2とPTM2)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge9.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge9.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(7, 10, 8, 11) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 obj <- as.logical(tcc$stat$q.value < param_FDR)#条件を満たすかどうかを判定した結果をobjに格納 points(result$a.value[obj], result$m.value[obj],#objがTRUEとなる点をハイライト pch=20, cex=1.5, col="magenta")#objがTRUEとなる点をハイライト legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題10は以下と同じ。13, 16, 14, 17列目のデータのみ抽出して、 アカゲザル2サンプル(G1群:RMF1とRMM1) vs. アカゲザル2サンプル(G2群:RMF2とRMM2)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge10.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge10.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(13, 16, 14, 17) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 obj <- as.logical(tcc$stat$q.value < param_FDR)#条件を満たすかどうかを判定した結果をobjに格納 points(result$a.value[obj], result$m.value[obj],#objがTRUEとなる点をハイライト pch=20, cex=1.5, col="magenta")#objがTRUEとなる点をハイライト legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題11は以下と同じ。13, 16, 15, 18列目のデータのみ抽出して、 アカゲザル2サンプル(G1群:RMF1とRMM1) vs. アカゲザル2サンプル(G2群:RMF3とRMM3)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge11.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge11.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(13, 16, 15, 18) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 obj <- as.logical(tcc$stat$q.value < param_FDR)#条件を満たすかどうかを判定した結果をobjに格納 points(result$a.value[obj], result$m.value[obj],#objがTRUEとなる点をハイライト pch=20, cex=1.5, col="magenta")#objがTRUEとなる点をハイライト legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題12は以下と同じ。1, 4, 13, 16列目のデータのみ抽出して、 ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の2群間比較を行っています。 q.value = 1の"ENSG00000125844"と "ENSG00000115325"をハイライトさせ、明らかにDEGでないものがどのあたりに位置するのかを示しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge12.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge12.png" #出力ファイル名を指定してout_f2に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(400, 310) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) param_geneid <- c("ENSG00000125844", "ENSG00000115325")#ハイライトさせたいgene_IDを指定 param_col <- "skyblue" #色を指定 param_cex <- 2 #点の大きさを指定(2なら通常の2倍、0.5なら通常の0.5倍) param_pch <- 20 #点の形状を指定(詳細はこちらとか) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 obj <- is.element(tcc$gene_id, param_geneid)#条件を満たすかどうかを判定した結果をobjに格納(param_geneidで指定した位置をTRUE) points(result$a.value[obj], result$m.value[obj],#条件を満たす点をハイライト pch=param_pch, cex=param_cex, col=param_col)#条件を満たす点をハイライト dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題13は以下と同じ。デフォルトのparam_subsetは、1, 4, 13, 16列目のデータのみ抽出して、 ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の2群間比較を行っていますが、適宜変更してください。 「FDR閾値を満たすもの」と「fold-change閾値を満たすもの」それぞれのM-A plotを作成しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge13.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge13_FDR.png" #出力ファイル名を指定してout_f2に格納 out_f3 <- "hoge13_FC.png" #出力ファイル名を指定してout_f3に格納 param_subset <- c(1, 4, 13, 16) #取り扱いたいサブセット情報を指定(HS1 vs. RM1) #param_subset <- c(1, 4, 7, 10) #取り扱いたいサブセット情報を指定(HS1 vs. PT1) #param_subset <- c(1, 4, 2, 5) #取り扱いたいサブセット情報を指定(HS1 vs. HS2) #param_subset <- c(7, 10, 8, 11) #取り扱いたいサブセット情報を指定(PT1 vs. PT2) #param_subset <- c(13, 16, 14, 17) #取り扱いたいサブセット情報を指定(RM1 vs. RM2) #param_subset <- c(13, 16, 15, 18) #取り扱いたいサブセット情報を指定(RM1 vs. RM3) param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 2 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_FC <- 2 #fold-change閾値(param_FC倍)を指定 param_fig <- c(400, 310) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_mar <- c(4, 4, 0, 0) #下、左、上、右の順で余白を指定(単位は行) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot; FDR) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 cex=0.8, cex.lab=1.2, #閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20, cex=1.2)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR < 0.05を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR < 0.10を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR < 0.20を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR < 0.30を満たす遺伝子数を表示 #ファイルに保存(M-A plot; fold-change) M <- getResult(tcc)$m.value #M-A plotのM値を抽出 hoge <- rep(1, length(M)) #初期値を1にしたベクトルhogeを作成 hoge[abs(M) > log2(param_FC)] <- 2 #条件を満たす位置に2を代入 cols <- c("black", "magenta") #色情報を指定してcolsに格納 png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=param_mar) #余白を指定 plot(tcc, col=cols, col.tag=hoge, xlim=c(-2, 17), ylim=c(-10.0, 11.5),#M-A plotを描画 cex=0.8, cex.lab=1.2, #閾値を満たすDEGをマゼンタ色にして描画 cex.axis=1.2, main="", #閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(", param_FC, "-fold)", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20)#凡例を作成 dev.off() #おまじない sum(abs(M) > log2(16)) #16倍以上発現変動する遺伝子数を表示 sum(abs(M) > log2(8)) #8倍以上発現変動する遺伝子数を表示 sum(abs(M) > log2(4)) #4倍以上発現変動する遺伝子数を表示 sum(abs(M) > log2(2)) #2倍以上発現変動する遺伝子数を表示
ReCountのgiladデータ(gilad_count_table.txt)で反復あり2群間比較を行う。 「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | TCC(Sun_2013)」例題1をベースに作成。 M-A plot周辺はオプションなどを多少変更しています。
in_f <- "gilad_count_table.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge1.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge1.png" #出力ファイル名を指定してout_f2に格納 param_G1 <- 3 #G1群のサンプル数を指定 param_G2 <- 3 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), tcc$count, result)#入力データの右側にDEG検出結果を結合したものをtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #余白を指定 plot(tcc, FDR=param_FDR, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
ReCountのmaqcデータ( maqc_count_table.txtと maqc_phenodata.txt) を入力としてサンプル間クラスタリング。
in_f1 <- "maqc_count_table.txt" #入力ファイル名を指定してin_f1に格納(カウントデータ) in_f2 <- "maqc_phenodata.txt" #入力ファイル名を指定してin_f2に格納(サンプルラベル情報) out_f <- "hoge.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(600, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") phenotype #確認してるだけです colnames(data) <- phenotype$tissue #dataオブジェクトの列名を変更 #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=1.2,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height") #樹形図(デンドログラム)の表示 dev.off() #おまじない
「解析 | クラスタリング | サンプル間 | TCC(Sun_2013)」の例題7をベースに作成。 入力は、20,689 genes×36 samplesのカウントデータ(sample_blekhman_36.txt)です。 縦軸の範囲を[0, 0.28]にするやり方を誰か教えて下さいm(_ _)m (「○○氏提供情報」とさせていただきます。)
in_f <- "sample_blekhman_36.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge7.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(700, 400) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_yrange <- c(0, 0.28) #表示したい縦軸の範囲を指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #本番 out <- clusterSample(data, dist.method="spearman",#クラスタリング実行結果をoutに格納 hclust.method="average", unique.pattern=TRUE)#クラスタリング実行結果をoutに格納 #ファイルに保存 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(0, 4, 1, 0)) #下、左、上、右の順で余白(行)を指定 plot(out, sub="", xlab="", cex.lab=1.2,#樹形図(デンドログラム)の表示 cex=1.3, main="", ylab="Height", #樹形図(デンドログラム)の表示 ylim=param_yrange) #樹形図(デンドログラム)の表示 dev.off() #おまじない
ReCountのmaqcデータ( maqc_count_table.txt)で反復あり2群間比較を行う。 「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | TCC(Sun_2013)」例題1をベースに作成。 M-A plot周辺はオプションなどを多少変更しています。
in_f <- "maqc_count_table.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge1.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge1.png" #出力ファイル名を指定してout_f2に格納 param_G1 <- 7 #G1群のサンプル数を指定 param_G2 <- 7 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(430, 350) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 sum(tcc$stat$q.value < param_FDR) #条件を満たす遺伝子数を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), tcc$count, result)#入力データの右側にDEG検出結果を結合したものをtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #余白を指定 plot(tcc, FDR=param_FDR, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
sum(tcc$stat$q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 7618*(1 - 0.05) 7843*(1 - 0.10) 8114*(1 - 0.20) 8338*(1 - 0.30)
technical replicatesのデータを足すだけ。
in_f <- "maqc_count_table.txt" #入力ファイル名を指定してin_fに格納 param_G1 <- 7 #G1群のサンプル数を指定 param_G2 <- 7 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 head(data[, data.cl == 1], n=3) #最初の3行分を表示(G1群) head(data[, data.cl == 2], n=3) #最初の3行分を表示(G2群) #本番(マージ) data_G1 <- rowSums(data[, data.cl == 1])#G1群をマージした結果をdata_G1に格納 data_G2 <- rowSums(data[, data.cl == 2])#G2群をマージした結果をdata_G2に格納 data <- cbind(data_G1, data_G2) #列方向で結合した結果をdataに格納 dim(data) #行数と列数を表示 head(data) #最初の6行分を表示 dim(unique(data)) #ユニークな発現パターン数を表示 sum(rowSums(data) > 0) #どこかのサンプルで1カウント分だけでも発現している遺伝子数を表示
getwd() list.files(pattern="maqc_pool")
「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC(Sun_2013)」 の例題5は以下と同じ。ReCountのmaqc (pooled)データ( maqc_pooledreps_count_table.txt)です。 52,580 genes×2 samplesのカウントデータ(G1群1サンプル vs. G2群1サンプル)です。
in_f <- "maqc_pooledreps_count_table.txt"#入力ファイル名を指定してin_fに格納 out_f1 <- "hoge5.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge5.png" #出力ファイル名を指定してout_f2に格納 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(400, 380) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #ファイルに保存(M-A plot) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 par(mar=c(4, 4, 0, 0)) #余白を指定 plot(tcc, FDR=param_FDR, main="", #param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画 xlab="A = (log2(G2) + log2(G1))/2",#閾値を満たすDEGをマゼンタ色にして描画 ylab="M = log2(G2) - log2(G1)") #閾値を満たすDEGをマゼンタ色にして描画 legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),#凡例を作成 col=c("magenta", "black"), pch=20)#凡例を作成 dev.off() #おまじない sum(tcc$stat$q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(tcc$stat$q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
TCCパッケージ(Sun et al., BMC Bioinformatics, 2013)のやり方です。
######################################## ### 反復あり(7 technical replicates) ######################################## in_f <- "maqc_count_table.txt" #入力ファイル名を指定してin_fに格納 param_G1 <- 7 #G1群のサンプル数を指定 param_G2 <- 7 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #TCC実行 library(TCC) #パッケージの読み込み tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 tcc <- estimateDE(tcc, test.method="edger")#DEG検出を実行した結果をtccに格納 p.value <- tcc$stat$p.value #p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_TCC_ari <- ranking #このランキング結果をranking_TCC_ariに格納 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示 ######################################## ### 反復なし(1 biological replicates; pooled) ######################################## in_f <- "maqc_pooledreps_count_table.txt"#入力ファイル名を指定してin_fに格納 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #TCC実行 library(TCC) #パッケージの読み込み tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 tcc <- estimateDE(tcc, test.method="deseq2")#DEG検出を実行した結果をtccに格納 p.value <- tcc$stat$p.value #p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_TCC_nasi <- ranking #このランキング結果をranking_TCC_nasiに格納 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示
edgeRパッケージ(Robinson et al., Bioinformatics, 2010)のやり方です。
######################################## ### 反復あり(7 technical replicates) ######################################## in_f <- "maqc_count_table.txt" #入力ファイル名を指定してin_fに格納 param_G1 <- 7 #G1群のサンプル数を指定 param_G2 <- 7 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data <- as.matrix(data) #データの型をmatrixにしている data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #edgeR実行 library(edgeR) #パッケージの読み込み d <- DGEList(counts=data, group=data.cl)#DGEListオブジェクトを作成してdに格納 d <- calcNormFactors(d) #TMM正規化を実行 d <- estimateCommonDisp(d) #the quantile-adjusted conditional maximum likelihood (qCML)法でcommon dispersionを計算している d <- estimateTagwiseDisp(d) #the quantile-adjusted conditional maximum likelihood (qCML)法でmoderated tagwise dispersionを計算している out <- exactTest(d) #exact test (正確確率検定)で発現変動遺伝子を計算した結果をoutに格納 p.value <- out$table$PValue #p-valueをp.valueに格納 ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_edgeR_ari <- ranking #このランキング結果をranking_edgeR_ariに格納 q.value <- p.adjust(p.value, method="BH")#q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示 ######################################## ### 反復なし(1 biological replicates; pooled) ######################################## in_f <- "maqc_pooledreps_count_table.txt"#入力ファイル名を指定してin_fに格納 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data <- as.matrix(data) #データの型をmatrixにしている data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #edgeR実行 library(edgeR) #パッケージの読み込み d <- DGEList(counts=data, group=data.cl)#DGEListオブジェクトを作成してdに格納 d <- calcNormFactors(d) #TMM正規化を実行 d <- estimateGLMCommonDisp(d, method="deviance", robust=TRUE, subset=NULL)#モデル構築 out <- exactTest(d) #exact test (正確確率検定)で発現変動遺伝子を計算した結果をoutに格納 p.value <- out$table$PValue #p-valueをp.valueに格納 ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_edgeR_nasi <- ranking #このランキング結果をranking_edgeR_nasiに格納 q.value <- p.adjust(p.value, method="BH")#q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示
DESeq2パッケージ(Love et al., Genome Biol., 2014)のやり方です。
######################################## ### 反復あり(7 technical replicates) ######################################## in_f <- "maqc_count_table.txt" #入力ファイル名を指定してin_fに格納 param_G1 <- 7 #G1群のサンプル数を指定 param_G2 <- 7 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data <- as.matrix(data) #データの型をmatrixにしている data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #DESeq2実行 library(DESeq2) #パッケージの読み込み colData <- data.frame(condition=as.factor(data.cl))#condition列にクラスラベル情報を格納したcolDataオブジェクトを作成 d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)#DESeqDataSetオブジェクトdの作成 d <- DESeq(d) #DESeq2を実行 tmp <- results(d) #実行結果を抽出 p.value <- tmp$pvalue ##p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_DESeq2_ari <- ranking #このランキング結果をranking_DESeq2_ariに格納 q.value <- tmp$padj #adjusted p-valueをq.valueに格納 q.value[is.na(q.value)] <- 1 #NAを1に置換している #q.value <- p.adjust(p.value, method="BH")#q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示 ######################################## ### 反復なし(1 biological replicates; pooled) ######################################## in_f <- "maqc_pooledreps_count_table.txt"#入力ファイル名を指定してin_fに格納 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data <- as.matrix(data) #データの型をmatrixにしている data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #DESeq2実行 library(DESeq2) #パッケージの読み込み colData <- data.frame(condition=as.factor(data.cl))#condition列にクラスラベル情報を格納したcolDataオブジェクトを作成 d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)#DESeqDataSetオブジェクトdの作成 d <- DESeq(d) #DESeq2を実行 tmp <- results(d) #実行結果を抽出 p.value <- tmp$pvalue ##p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_DESeq2_nasi <- ranking #このランキング結果をranking_DESeq2_nasiに格納 q.value <- tmp$padj #adjusted p-valueをq.valueに格納 q.value[is.na(q.value)] <- 1 #NAを1に置換している #q.value <- p.adjust(p.value, method="BH")#q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示
DESeq2パッケージ (Love et al., Genome Biol., 2014)のやり方です。 DESeq2オリジナルのq-valueではなく、p-valueからp.adjust関数を用いてq.valueを計算しています。
######################################## ### 反復あり(7 technical replicates) ######################################## in_f <- "maqc_count_table.txt" #入力ファイル名を指定してin_fに格納 param_G1 <- 7 #G1群のサンプル数を指定 param_G2 <- 7 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data <- as.matrix(data) #データの型をmatrixにしている data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #DESeq2実行 library(DESeq2) #パッケージの読み込み colData <- data.frame(condition=as.factor(data.cl))#condition列にクラスラベル情報を格納したcolDataオブジェクトを作成 d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)#DESeqDataSetオブジェクトdの作成 d <- DESeq(d) #DESeq2を実行 tmp <- results(d) #実行結果を抽出 p.value <- tmp$pvalue ##p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_DESeq2_ari <- ranking #このランキング結果をranking_DESeq2_ariに格納 #q.value <- tmp$padj #adjusted p-valueをq.valueに格納 #q.value[is.na(q.value)] <- 1 #NAを1に置換している q.value <- p.adjust(p.value, method="BH")#q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示 ######################################## ### 反復なし(1 biological replicates; pooled) ######################################## in_f <- "maqc_pooledreps_count_table.txt"#入力ファイル名を指定してin_fに格納 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data <- as.matrix(data) #データの型をmatrixにしている data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 #DESeq2実行 library(DESeq2) #パッケージの読み込み colData <- data.frame(condition=as.factor(data.cl))#condition列にクラスラベル情報を格納したcolDataオブジェクトを作成 d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)#DESeqDataSetオブジェクトdの作成 d <- DESeq(d) #DESeq2を実行 tmp <- results(d) #実行結果を抽出 p.value <- tmp$pvalue ##p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 ranking_DESeq2_nasi <- ranking #このランキング結果をranking_DESeq2_nasiに格納 #q.value <- tmp$padj #adjusted p-valueをq.valueに格納 #q.value[is.na(q.value)] <- 1 #NAを1に置換している q.value <- p.adjust(p.value, method="BH")#q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示
第1部 | 統計解析 | トランスクリプトーム解析2(2016.07.22)
- 講義資料PDF(2016.07.13版; 約7MB)
- hoge.zip(2016.06.02版; 約1MB)
- 3群間比較用データ:Blekhman et al., Genome Res., 2010 (スライド3)
- 性能評価論文(3群間比較用):Tang et al., BMC Bioinformatics, 2015 (スライド4)
- データ正規化周辺 (スライド5)
- RPM正規化(RPKMの一部):Mortazavi et al., Nat Methods, 2008
- TMM正規化:edgeR:Robinson et al., Bioinformatics, 2010
- TbT正規化(後にTCCに実装):Kadota et al., Algorithms Mol. Biol., 2012
- iDEGES正規化:TCC:Sun et al., BMC Bioinformatics, 2013
- 3群間比較(反復あり) (スライド9)
- DEG数の見積もり (スライド13)
- オプション明示(ANOVA) (スライド29)
- デザイン行列 (スライド31)
- 帰無仮説(G1 = G2) (スライド42)
- 帰無仮説(G1 = G3) (スライド52)
- 帰無仮説(G2 = G3) (スライド66)
- コントラストでG1 = G2 (スライド77)
- コントラストでG1 = G3 (スライド78)
- HS vs. PT (2群間比較) (スライド85)
- HS vs. RM (2群間比較) (スライド86)
- PT vs. RM (2群間比較) (スライド87)
- 遺伝子クラスタリング(MBCluster.Seq) (スライド97)
- TCC正規化を合わせる (スライド107)
- TCC発現変動とも合体 (スライド111)
- 割合で表示したい場合 (スライド120のおまけ)
- baySeqで3群間比較 (スライド124)
- 性能評価論文のTable 4 (スライド132)
- TCCのq-valueを... (スライド143)
- 反復なし3群間比較(TCC) (スライド152)
- 反復なし3群間比較(DESeq2) (スライド155)
20,689 genes×18 samplesのカウントデータ(sample_blekhman_18.txt)です。
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | TCC(Sun_2013)」 の例題7は以下と同じ。サンプルデータ42のリアルデータ(sample_blekhman_18.txt)です。 20,689 genes×18 samplesのカウントデータで、ヒト(HS)、チンパンジー(PT)、アカゲザル(RM)の3生物種間比較です (Blekhman et al., Genome Res., 2010)。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge7.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
7236*(1 - 0.05)
8485*(1 - 0.10)
10068*(1 - 0.20)
11467*(1 - 0.30)
packageVersion("TCC") #パッケージのバージョン情報を表示
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」 の例題1をベースに作成。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge1.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_coef <- c(2, 3) #デザイン行列中の除きたい列を指定(reduced model作成用) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) design <- model.matrix(~ as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,#DEG検出を実行した結果をtccに格納 design=design, coef=param_coef)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 design #デザイン行列の情報を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
data.cl
as.factor(data.cl)
unique(data.cl)
design
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」 の例題2をベースに作成。「G1群 vs. G2群」の比較を行いたいときの指定方法(帰無仮説: G1 = G2)です。 「full modelに相当するデザイン行列designの2列目のパラメータを除いたものをreduced modelとする」という指定に相当します。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge2.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_coef <- 2 #デザイン行列中の除きたい列を指定(reduced model作成用) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) design <- model.matrix(~ as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,#DEG検出を実行した結果をtccに格納 design=design, coef=param_coef)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 design #デザイン行列の情報を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」 の例題3をベースに作成。「G1群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G1 = G3)です。 「full modelに相当するデザイン行列designの3列目のパラメータを除いたものをreduced modelとする」という指定に相当します。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge3.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_coef <- 3 #デザイン行列中の除きたい列を指定(reduced model作成用) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) design <- model.matrix(~ as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,#DEG検出を実行した結果をtccに格納 design=design, coef=param_coef)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 design #デザイン行列の情報を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」 の例題4をベースに作成。「G2群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G2 = G3)です。 param_coefの枠組みではうまくG2 vs. G3をうまく表現できないので、コントラストという別の枠組みで指定しています。 思考回路としては、「a1*G1 + a2*G2 + a3*G3 = 0」として、この場合の目的である「G2 = G3という帰無仮説」を作成できるように、係数a1, a2, a3に適切な数値を代入することです。 ここではa1 = 0、a2 = -1, a3 = 1にすることで目的の帰無仮説を作成できます。 以下ではc(0, -1, 1)と指定していますが、 c(0, 1, -1)でも構いません。 理由はどちらでもG2 = G3を表現できているからです。 尚、full modelに相当するデザイン行列の作成手順も若干異なります。具体的には、model.matrix関数実行時に「0 + 」を追加しています。 これによって、最初の1列目が全て1になるようなG1群を基準にして作成したデザイン行列ではなく、各群が各列になるようにしています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge4.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_contrast <- c(0, -1, 1) #コントラスト情報を指定(reduced model作成用) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) #design <- model.matrix(~ as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 design <- model.matrix(~ 0 + as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,#DEG検出を実行した結果をtccに格納 design=design, contrast=param_contrast)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 design #デザイン行列の情報を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」 の例題5をベースに作成。コントラストで「G1群 vs. G2群」の比較を行いたいときの指定方法(帰無仮説: G1 = G2)です。 ここではa1 = 1、a2 = -1, a3 = 0にすることで目的の帰無仮説を作成できます。 以下ではc(1, -1, 0)と指定していますが、 c(-1, 1, 0)でも構いません。 理由はどちらでもG1 = G2を表現できているからです。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge5.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_contrast <- c(1, -1, 0) #コントラスト情報を指定(reduced model作成用) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) #design <- model.matrix(~ as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 design <- model.matrix(~ 0 + as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,#DEG検出を実行した結果をtccに格納 design=design, contrast=param_contrast)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 design #デザイン行列の情報を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」 の例題6をベースに作成。コントラストで「G1群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G1 = G3)です。 ここではa1 = 1、a2 = 0, a3 = -1にすることで目的の帰無仮説を作成できます。 以下ではc(1, 0, -1)と指定していますが、 c(-1, 0, 1)でも構いません。 理由はどちらでもG1 = G3を表現できているからです。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge6.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_contrast <- c(1, 0, -1) #コントラスト情報を指定(reduced model作成用) #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) #design <- model.matrix(~ as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 design <- model.matrix(~ 0 + as.factor(data.cl))#デザイン行列を作成した結果をdesignに格納 tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,#DEG検出を実行した結果をtccに格納 design=design, contrast=param_contrast)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 design #デザイン行列の情報を表示 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題7をベースに作成。1:6, 7:12 列目のデータのみ抽出し、通常の6 HS samples vs. 6 PT samplesの2群間比較を行っています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge_2_HS_PT.txt" #出力ファイル名を指定してout_f1に格納 param_subset <- c(1:6, 7:12) #取り扱いたいサブセット情報を指定 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題7をベースに作成。1:6, 13:18 列目のデータのみ抽出し、通常の6 HS samples vs. 6 RM samplesの2群間比較を行っています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge_2_HS_RM.txt" #出力ファイル名を指定してout_f1に格納 param_subset <- c(1:6, 13:18) #取り扱いたいサブセット情報を指定 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)」 の例題7をベースに作成。7:12, 13:18 列目のデータのみ抽出し、通常の6 PT samples vs. 6 RM samplesの2群間比較を行っています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge_2_PT_RM.txt" #出力ファイル名を指定してout_f1に格納 param_subset <- c(7:12, 13:18) #取り扱いたいサブセット情報を指定 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2))#G1群を1、G2群を2としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示
「解析 | クラスタリング | 遺伝子間(基礎) | MBCluster.Seq(Si_2014)」 の例題4と同じです。遺伝子ごとにparam_clust_numで指定したクラスターのどこに割り振られたかの情報を、 クラスターごとにソートして保存しています。とりあえずクラスター数を10にしています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge4.png" #出力ファイル名を指定してout_f1に格納(pngファイル) out_f2 <- "hoge4.txt" #出力ファイル名を指定してout_f2に格納(txtファイル) param_fig <- c(800, 500) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_clust_num <- 10 #クラスター数を指定 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 #必要なパッケージをロード library(MBCluster.Seq) #パッケージの読み込み #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 dim(data) #オブジェクトdataの行数と列数を表示 #前処理(基礎情報取得) hoge <- RNASeq.Data(data, Normalizer=NULL,#クラスタリングに必要な基礎情報を取得 Treatment=data.cl, GeneID=rownames(data))#クラスタリングに必要な基礎情報を取得 #本番(クラスタリング) c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,#K-means clusteringのクラスター中心の初期値を取得 model="nbinom", print.steps=F)#K-means clusteringのクラスター中心の初期値を取得 cls <- Cluster.RNASeq(data=hoge, model="nbinom",#クラスタリング centers=c0$centers, method="EM")#クラスタリング tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)#hybrid-hierarchical clustering(ツリー構造の構築) table(cls$cluster) #各クラスターに属する遺伝子数を表示 #ファイルに保存(pngファイル) png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)#描画 dev.off() #おまじない #ファイルに保存(txtファイル) tmp <- cbind(rownames(data), data, cls$cluster)#保存したい情報をtmpに格納 tmp <- tmp[order(cls$cluster),] #クラスター番号順にソートした結果をtmpに格納 write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)#tmpの中身をout_f2で指定したファイル名で保存
「解析 | クラスタリング | 遺伝子間(応用) | TCC正規化(Sun_2013)+MBCluster.Seq(Si_2014)」 の例題4は以下と同じ。TCC正規化後のデータを入力としてMBCluster.Seqを実行するやり方。 とりあえずクラスター数を5にしています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge4.png" #出力ファイル名を指定してout_f1に格納(pngファイル) out_f2 <- "hoge4.txt" #出力ファイル名を指定してout_f2に格納(txtファイル) param_fig <- c(800, 500) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_clust_num <- 5 #クラスター数を指定 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 #必要なパッケージをロード library(MBCluster.Seq) #パッケージの読み込み library(TCC) #パッケージの読み込み #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み dim(data) #オブジェクトdataの行数と列数を表示 #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 norm.factors <- tcc$norm.factors #TCC正規化係数をnorm.factorsに格納 ef.libsizes <- colSums(data)*norm.factors#総リード数(library sizes)にTCC正規化係数を掛けて得たeffective library sizes情報をef.libsizesに格納 size.factors <- ef.libsizes/mean(ef.libsizes)#effective library sizesを正規化してサイズファクターを得る #前処理(正規化) hoge <- RNASeq.Data(data, Normalizer=size.factors,#クラスタリングに必要な基礎情報を取得 Treatment=data.cl, GeneID=rownames(data))#クラスタリングに必要な基礎情報を取得 #本番(クラスタリング) c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,#K-means clusteringのクラスター中心の初期値を取得 model="nbinom", print.steps=F)#K-means clusteringのクラスター中心の初期値を取得 cls <- Cluster.RNASeq(data=hoge, model="nbinom",#クラスタリング centers=c0$centers, method="EM")#クラスタリング tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)#hybrid-hierarchical clustering(ツリー構造の構築) table(cls$cluster) #各クラスターに属する遺伝子数を表示 #ファイルに保存(pngファイル) png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)#描画 dev.off() #おまじない #ファイルに保存(txtファイル) normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 tmp <- cbind(rownames(data), normalized, cls$cluster)#保存したい情報をtmpに格納 tmp <- tmp[order(cls$cluster),] #クラスター番号順にソートした結果をtmpに格納 write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)#tmpの中身をout_f2で指定したファイル名で保存
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」 の例題9は以下と同じ。TCC正規化後のデータを入力としてMBCluster.Seqを実行するやり方。 さらにTCCによるANOVA的解析(どこかの群間で発現変動する遺伝子の検出)結果とも合わせている。 とりあえずクラスター数を10にしています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f1 <- "hoge9.txt" #出力ファイル名を指定してout_f1に格納(txtファイル) out_f2 <- "hoge9.png" #出力ファイル名を指定してout_f2に格納(pngファイル) param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_fig <- c(800, 500) #ファイル出力時の横幅と縦幅を指定(単位はピクセル) param_clust_num <- 10 #クラスター数を指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み library(MBCluster.Seq) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(TCCクラスオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(TCC正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 norm.factors <- tcc$norm.factors #TCC正規化係数をnorm.factorsに格納 ef.libsizes <- colSums(data)*norm.factors#総リード数(library sizes)にTCC正規化係数を掛けて得たeffective library sizes情報をef.libsizesに格納 size.factors <- ef.libsizes/mean(ef.libsizes)#effective library sizesを正規化してサイズファクターを得る #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 result <- getResult(tcc, sort=FALSE) #p値などの計算結果をresultに格納 #前処理(MBCluster.Seq基礎情報取得) hoge <- RNASeq.Data(data, Normalizer=size.factors,#クラスタリングに必要な基礎情報を取得 Treatment=data.cl, GeneID=rownames(data))#クラスタリングに必要な基礎情報を取得 #本番(MBCluster.Seqクラスタリング) set.seed(2016) #おまじない(同じ乱数になるようにするため) c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,#K-means clusteringのクラスター中心の初期値を取得 model="nbinom", print.steps=F)#K-means clusteringのクラスター中心の初期値を取得 cls <- Cluster.RNASeq(data=hoge, model="nbinom",#クラスタリング centers=c0$centers, method="EM")#クラスタリング tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)#hybrid-hierarchical clustering(ツリー構造の構築) #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, result, cls$cluster)#保存したい情報をtmpに格納 tmp <- tmp[order(tmp$rank),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 #ファイルに保存(pngファイル) png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)#描画 dev.off() #おまじない #後処理(各クラスターに属する遺伝子数を表示) table(cls$cluster) #各クラスターに属する遺伝子数を表示(全遺伝子) table(cls$cluster[q.value < 0.05]) #各クラスターに属する遺伝子数を表示(q-value < 0.05を満たす遺伝子のみ) table(cls$cluster[q.value < 0.10]) #各クラスターに属する遺伝子数を表示(q-value < 0.10を満たす遺伝子のみ) table(cls$cluster[q.value < 0.20]) #各クラスターに属する遺伝子数を表示(q-value < 0.20を満たす遺伝子のみ) table(cls$cluster[q.value < 0.30]) #各クラスターに属する遺伝子数を表示(q-value < 0.30を満たす遺伝子のみ)
割合で表示する基本形、および表示桁数を指定して表示するやり方です。
#基本形 table(cls$cluster)/length(q.value) table(cls$cluster[q.value < 0.05])/sum(q.value < 0.05) table(cls$cluster[q.value < 0.10])/sum(q.value < 0.10) table(cls$cluster[q.value < 0.20])/sum(q.value < 0.20) table(cls$cluster[q.value < 0.30])/sum(q.value < 0.30) #sprintfで小数点以下の桁数を2に限定 sprintf("%3.2f", table(cls$cluster)/length(q.value)) sprintf("%3.2f", table(cls$cluster[q.value < 0.05])/sum(q.value < 0.05)) sprintf("%3.2f", table(cls$cluster[q.value < 0.10])/sum(q.value < 0.10)) sprintf("%3.2f", table(cls$cluster[q.value < 0.20])/sum(q.value < 0.20)) sprintf("%3.2f", table(cls$cluster[q.value < 0.30])/sum(q.value < 0.30))
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | baySeq(Hardcastle_2010)」 の例題4は以下と同じ。param_samplesizeで指定するリサンプリング回数を500にして実行時間を大幅に短縮しています。約4分。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge4.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 6 #G1群のサンプル数を指定 param_G2 <- 6 #G2群のサンプル数を指定 param_G3 <- 6 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 param_samplesize <- 500 #ブートストラップリサンプリング回数(100000が推奨。大きい値ほど計算時間増加) #必要なパッケージをロード library(baySeq) #パッケージの読み込み #入力ファイルの読み込みとラベル情報の作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 #前処理(countDataオブジェクトの作成) NDE <- c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3))#non-DEGのパターンを作成 DE <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))#DEGのパターンを作成 ba <- new("countData", data=as.matrix(data), replicates=data.cl)#countDataオブジェクトを作成 groups(ba) <- list(NDE=NDE, DE=DE) #non-DEGとDEGのパターン情報を格納 #本番(正規化) libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")#edgeRパッケージ中の正規化法を実行 #本番(DEG検出) set.seed(2015) #おまじない(同じ乱数になるようにするため) ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)#事前分布を推定 ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)#事後確率を計算 res <- topCounts(ba, group="DE", normaliseData=T, number=nrow(data))#(発現変動順にソート済みの)解析結果を抽出 res <- res[rownames(data),] #解析結果をオリジナルの並びに再ソート q.value <- res$FDR.DE #FDR.DEの情報ををq.valueに格納 ranking <- rank(q.value) #q.valueでランキングした結果をrankingに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(res), res, ranking)#保存したい情報をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.20) #FDR = 0.20 (q-value < 0.20)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 #結果のまとめ(全遺伝子) head(res$ordering) #最初の6遺伝子分のパターンを表示 table(res$ordering) #パターンごとの出現頻度を表示 table(res$ordering)/length(res$ordering)#パターンごとの出現確率を表示 #結果のまとめ(FDR閾値を満たすものを概観) obj <- (q.value < param_FDR) #条件を満たすかどうかを判定した結果をobjに格納 sum(obj) #条件を満たす数を表示 table(res$ordering[obj]) #パターンごとの出現頻度を表示(条件を満たすもののみ) table(res$ordering[obj])/length(res$ordering[obj])#パターンごとの出現確率を表示(条件を満たすもののみ)
Tang et al., BMC Bioinformacis, 2015のTable 4への直接リンクはないようなので、
ここから辿ってください。
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | TCC(Sun_2013)」 の例題7をベースにして、必要最小限のコードを実行。baySeq実行結果のres$ordering情報を利用した発現パターン分類を行う。
#必要なパッケージをロード library(TCC) #パッケージの読み込み #前処理(TCCクラスオブジェクトの作成) tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",#正規化を実行した結果をtccに格納 iteration=3, FDR=0.1, floorPDEG=0.05)#正規化を実行した結果をtccに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 #結果のまとめ(FDR閾値を満たすものを概観) obj <- (tcc$stat$q.value < param_FDR)#条件を満たすかどうかを判定した結果をobjに格納 sum(obj) #条件を満たす数を表示 table(res$ordering[obj]) #パターンごとの出現頻度を表示(条件を満たすもののみ) table(res$ordering[obj])/length(res$ordering[obj])#パターンごとの出現確率を表示(条件を満たすもののみ)
「解析 | 発現変動 | 3群間 | 対応なし | 複製なし | TCC(Sun_2013)」 の例題4は以下と同じ。サンプルデータ42の20,689 genes×18 samplesのリアルカウントデータ(sample_blekhman_18.txt)です。 1, 7, 13列目のデータのみ抽出して、 反復なし3群間比較(HSF1 vs. PTF1 vs. RMF1)としています。正規化後のデータで発現変動順にソートした結果を出力しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge4.txt" #出力ファイル名を指定してout_fに格納 param_subset <- c(1, 7, 13) #取り扱いたいサブセット情報を指定 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 param_G3 <- 1 #G3群のサンプル数を指定 param_FDR <- 0.05 #false discovery rate (FDR)閾値を指定 #必要なパッケージをロード library(TCC) #パッケージの読み込み #入力ファイルの読み込み data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み #前処理(サブセットの抽出とTCCクラスオブジェクトの作成) data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 tcc <- new("TCC", data, data.cl) #TCCクラスオブジェクトtccを作成 colnames(data) #列名を表示 #本番(正規化) tcc <- calcNormFactors(tcc, norm.method="deseq2",#正規化を実行した結果をtccに格納 test.method="deseq2", iteration=3)#正規化を実行した結果をtccに格納 normalized <- getNormalizedData(tcc) #正規化後のデータを取り出してnormalizedに格納 #本番(DEG検出) tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)#DEG検出を実行した結果をtccに格納 p.value <- tcc$stat$p.value #p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 q.value <- tcc$stat$q.value #q-valueをq.valueに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(tcc$count), normalized, p.value, q.value, ranking)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(ranking),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示
「解析 | 発現変動 | 3群間 | 対応なし | 複製なし | DESeq2(Love_2014)」 の例題2は以下と同じ。サンプルデータ42の20,689 genes×18 samplesのリアルカウントデータ(sample_blekhman_18.txt)です。 1, 7, 13列目のデータのみ抽出して、 反復なし3群間比較(HSF1 vs. PTF1 vs. RMF1)としています。正規化後のデータで発現変動順にソートした結果を出力しています。
in_f <- "sample_blekhman_18.txt" #入力ファイル名を指定してin_fに格納 out_f <- "hoge2.txt" #出力ファイル名を指定してout_fに格納 param_subset <- c(1, 7, 13) #取り扱いたいサブセット情報を指定 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 param_G3 <- 1 #G3群のサンプル数を指定 #必要なパッケージをロード library(DESeq2) #パッケージの読み込み #入力ファイルの読み込みとサブセットの作成 data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")#in_fで指定したファイルの読み込み data <- data[,param_subset] #param_subsetで指定した列の情報のみ抽出 #前処理(DESeqDataSetオブジェクトの作成) data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))#G1群を1、G2群を2、G3群を3としたベクトルdata.clを作成 colData <- data.frame(condition=as.factor(data.cl))#condition列にクラスラベル情報を格納したcolDataオブジェクトを作成 d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)#DESeqDataSetオブジェクトdの作成 #本番(DEG検出) d <- DESeq(d) #DESeq2を実行 #d <- estimateSizeFactors(d) #正規化を実行した結果をdに格納 ##sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))#size factorsをさらに正規化(TCCは内部的にこれを行っている) #d <- estimateDispersions(d) #モデル構築(ばらつきの程度を見積もっている) #d <- nbinomLRT(d, full= ~condition, reduced= ~1)#検定 normalized <- counts(d, normalized=T) #正規化後のデータを取り出してnormalizedに格納 tmp <- results(d) #実行結果を抽出 p.value <- tmp$pvalue ##p-valueをp.valueに格納 p.value[is.na(p.value)] <- 1 #NAを1に置換している q.value <- tmp$padj #adjusted p-valueをq.valueに格納 q.value[is.na(q.value)] <- 1 #NAを1に置換している ranking <- rank(p.value) #p.valueでランキングした結果をrankingに格納 #ファイルに保存(テキストファイル) tmp <- cbind(rownames(data), normalized, p.value, q.value, ranking)#正規化後のデータの右側にDEG検出結果を結合したものをtmpに格納 tmp <- tmp[order(ranking),] #発現変動順にソートした結果をtmpに格納 write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)#tmpの中身を指定したファイル名で保存 #様々なFDR閾値を満たす遺伝子数を表示 sum(q.value < 0.05) #FDR = 0.05 (q-value < 0.05)を満たす遺伝子数を表示 sum(q.value < 0.10) #FDR = 0.10 (q-value < 0.10)を満たす遺伝子数を表示 sum(q.value < 0.30) #FDR = 0.30 (q-value < 0.30)を満たす遺伝子数を表示 sum(q.value < 0.50) #FDR = 0.50 (q-value < 0.50)を満たす遺伝子数を表示 sum(q.value < 0.70) #FDR = 0.70 (q-value < 0.70)を満たす遺伝子数を表示 sum(q.value < 0.80) #FDR = 0.80 (q-value < 0.80)を満たす遺伝子数を表示 sum(p.adjust(p.value, method="BH") < 0.05)#FDR閾値(q.value < 0.05)を満たす遺伝子数を表示(TCCはp-valueをもとにBH法でq-value情報を得ている) sum(p.adjust(p.value, method="BH") < 0.10)#FDR閾値(q.value < 0.10)を満たす遺伝子数を表示(TCCはp-valueをもとにBH法でq-value情報を得ている) sum(p.adjust(p.value, method="BH") < 0.30)#FDR閾値(q.value < 0.30)を満たす遺伝子数を表示(TCCはp-valueをもとにBH法でq-value情報を得ている) sum(p.adjust(p.value, method="BH") < 0.50)#FDR閾値(q.value < 0.50)を満たす遺伝子数を表示(TCCはp-valueをもとにBH法でq-value情報を得ている) sum(p.adjust(p.value, method="BH") < 0.70)#FDR閾値(q.value < 0.70)を満たす遺伝子数を表示(TCCはp-valueをもとにBH法でq-value情報を得ている) sum(p.adjust(p.value, method="BH") < 0.80)#FDR閾値(q.value < 0.80)を満たす遺伝子数を表示(TCCはp-valueをもとにBH法でq-value情報を得ている)
第2部 | NGS解析(初~中級) | について
第2部は、Bio-Linux環境で行います。ほぼ完成です(2016年07月20日現在)。2016年07月20日に講義資料PDFをアップロードしました。 2016年07月21日にコピペ用コマンド集も公開しました。
- 予習事項(必須)
- Bio-Linux8(第2部で利用するovaファイル)の導入
- 共有フォルダ設定確認
- Win:「C:\Users\株式会社\Desktop\share」を変更
- Mac:「C:\Users\株式会社\Desktop\share」を変更
- Bio-Linux環境での作業に一通り慣れておく。Bio-Linux特有の不具合への対処法を身につけておきましょう。
- 平成27年度の「Bio-Linux 8とRのインストール状況確認(7月22日分)」のBio-Linuxに関する41項目のチェックリストをクリア
- (第4回だけでなく第2-4回のウェブ資料PDFにも目を通している)
- ゲストOS、ホストOSなどと言われてうろたえない(第1回)
- Linux環境では、一般にダブルクリックでソフトウェアのインストールはしない(第1回)
- Perl, Ruby, Pythonはプログラミング言語である(第1回)
- 「円(yen)マーク」と「バックスラッシュ」の違いは見栄えの問題であり、文字コードとしては同じなので気にしない(第2回)
- コマンドとオプションの間には「スペース」が入るものだ(第2回)
- .(どっと)から始まるファイル名のもの(の多く)は、環境設定ファイルである(第2回)
- 全角文字をコマンドとして打ち込むのは非常識である(第2回)
- ファイルの拡張子は常識的に使う。例えばFASTQ形式ファイルの拡張子を.fastaや.faなどにしない(第2回)
- 「Tabキーによる補完」、「上下左右の矢印キー」を駆使して効率的にコマンドを打てるようになっている(第2回)
- リダイレクトやパイプを含むコマンドを眺めて、何をやっているものかが大体わかるようになっている(第2回)
- Bio-Linuxの左側の引き出しみたいなアイコンまたはターミナル上で、 ホスト <--> ゲスト間のファイルのやり取りが自在にできる(第3回)
- ホストからゲストへはドラッグ&ドロップ可能だが、ゲストからホストへはドラッグ&ドロップ不可能[W8-1](第3回)
- ターミナルの画面サイズや不具合は自分で変更および対処可能[W9-2-4](第3回)
- 共有フォルダの概念を理解している[W9-3](第3回)
- rmコマンドは「ゴミ箱」への移動ではなく本当に消える[W10-4](第3回)
- ダウンロードしたいファイルのURL情報取得ができる[W11-1](第3回)
- ゲストOSのネットワーク接続が不調の場合は、つながるまで待つのではなく、ホストOS上で必要なファイルをダウンロードして共有フォルダ 経由でゲストOS上の適切な場所に置くことができる[W11-1](第3回)
- 圧縮・解凍コマンド(zip, unzip, gzip, gunzip, bzip2, and bunzip2)の使い方をマスターしている[W13](第3回)
- headやtailコマンドでファイル中の最初と最後の部分を確認することができる[W14](第3回)
- cat, more, lessコマンドの基本的な使用法(脱出したいときはCTRL + Cなど)を理解している[W14-4からW14-6](第3回)
- 多くのLinuxコマンドには様々なオプションがあり、--hや--helpなどで利用可能なオプションを眺めることができる[W15-2](第3回)
- PDFファイルやPowerPointファイル中のコマンドをコピペするときは気をつける[W16-1](第3回)
- コマンドオプション利用時の「ハイフン」と「ダッシュ」の気をつける[W16-3](第3回)
- 初心者向けテキストエディタgeditの基本的な利用法は知っている[W17](第3回)
- 仮想環境全体のディスク使用状況把握(dfコマンド)や、カレントディレクトリのディスク使用量(duコマンド)を把握し、 適切に不要なファイルを削除できる[W18-2からW18-4](第3回)
- 「.(どっと)」はカレントディレクトリを表し、「..(どっとどっと)」は1つ上の階層のディレクトリを意味する[W18-3](第3回)
- 複数のコマンドを組み合わせたパイプ(|)利用の基本を知っている[W19](第3回)
- 連載原稿中では乳酸菌RNA-seqデータ(SRR616268)を例に解説しているが、 NGSハンズオン講習会(または予習)では、(自己責任で)自分が興味あるデータで平行して解析してもよいことを知っている[W20以降](第3回)
- bzip2はgzipに比べて圧縮率は高く、圧縮に要する時間も早いが、解凍は遅い[W22-5からW22-6](第3回)
- ファイルサイズが大きいものは、一つ一つの処理に要する時間がとてつもなく長くなることを知っている[W23](第3回)
- コマンドオプション、パイプ、リダイレクトを駆使することでNGSデータのサブセットを抽出できることを知っている[W25-2](第3回)
- アスタリスク(*)などのワイルドカードというものがあり、それらの挙動がある程度わかる[W1-2からW1-3](第4回)
- ゲストOSがフリーズ状態に遭遇しても、自力で再起動するなりして復旧できる[W2-1](第4回)
- Permission問題はchmodで自力で解決できる[W3-1](第4回)
- 同じ目的を達成する上で様々なやり方があることを知っている[W3-2](第4回)
- ファイルのダウンロード時に拡張子がオリジナルと変わっていることがある[W4-4](第4回)
- 自動マウント(オートマウント)の設定ができている[W4-5; W4-7](第4回)
- 絶対パス、相対パスの概念が理解できている[W4-6](第4回)
- 文字コード(改行コード)をodやfileコマンドで確認し、perlで変換できる[W5-2; W5-3](第4回)
- 「ls」が「3s」になったときは、「Shift + Numlk」を押すと元に戻る。 PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB)の項目7にあります。
- 「*」が「"」になったときは、おそらく英語キーボードを表す「En」になっている。 これを日本語キーボードを表す「Ja」押すと打てるようになる。 PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB)の項目7にあります。
- 日本乳酸菌学会誌連載第6回のウェブ資料として提供しているovaファイルからの導入手順を予習
- ovaファイルの導入手順:Windows用(2015.12.28版; 約2MB)
- ovaファイルの導入手順:Macintosh用(2015.12.28版; 約2MB)
- 予習事項(推奨)
- 2016年07月25日分(NGS解析基礎)の講義資料PDF(2016.08.03版)
- 2016年07月26日分(Reseq)の講義資料PDF(2016.08.03版)
- 2016年07月27日分(RNA-seq)の講義資料PDF(2016.08.03版)
- 2016年07月28日分(ChIP-seq)の講義資料PDF(2016.08.03版)
- 予習事項(ほぼ任意)
- (Rで)塩基配列解析中の「書籍 | 日本乳酸菌学会誌 | について」の第4回分まで
- 平成27年度講習会の「Linux基礎(7月23日分)」の自習
- 平成27年度講習会の「NGS解析(8月3-4日分)」の自習
持込PCのヒトのみ。第2部で利用するovaファイル(6GB程度)を導入しておいてください(基本的には7/19中)。 中身は、乳酸菌NGS連載第4回終了時点のovaファイルをベースに、第2部でアメリエフさんが使うファイルやプログラムを追加したものとなる予定です。 準備ができ次第ダウンロードURLを提示します。
具体的には、キーバインド(キー配置)が勝手に変更されたり、ウィンドウ画面サイズを変更する際にしばしばホストOSが勝手に再起動してログイン画面に戻ってしますことなどです。 基本的な対処法はゲストOSの再起動です。ls, cd, pwd, lessなどの基本的なLinuxコマンドは使えるようになっておきましょう。
以下に示してありますので、そちらをご利用ください。昨年度のチェックリストとほぼ同じです。 この予習事項を重複する内容や予習事項にないコマンドなども出てくるかもしれません。予めご了承ください。 第2部に限っていえば、アメリエフさんが講師なのでやさしく丁寧に教えてくれます。 下記項目の予習内容が若干不十分でもどうにかなるとは思いますが、きっちり予習をしてきた他の受講生が不快に思わない程度にTAに助けを求めるよう心がけましょう。
予習事項1は、ここのやり方を参考にしてください(もちろん乳酸菌NGS連載第2回最後の「1. VirtualBox、および2. Extension Pack」のインストールが完了しているという前提です)。 持込PCのヒトも貸与PCのヒトも、PC関連情報PDF20160719_pcinfo_20160709.pdf(2016.07.09版; 約4MB)を一通り眺めておいてください。
参加予定の平成28年度講習会資料に目を通しておきましょう。随時差し替えます。
前述のように講習会で利用するovaファイルは、第4回終了時点のものをベースにアメリエフさんが作成予定です。 そのため、NGS連載第4回の最後まで一通り自習しておいたほうがいいと思います。
第3部 | NGS解析(中~上級) | について
多少変更するかもしれませんが、ほぼ完成です(2016年07月14日現在)。第3部は、主にBio-Linux環境で行います。 2016年7月14日に「農学生命情報科学特論II」受講者用の 課題(kadai_tokuronII.pdf)と仮想NGSデータ(kadai_20160720.fasta)を追加しました。 提出期限は8月30日です。
- 予習事項(必須)
- 第1部と第2部の予習事項内容
- 共有フォルダ設定確認(上記予習事項に含まれるが念のため)
- Win:「C:\Users\iu\Desktop\share」を変更
- Mac:「C:\Users\iu\Desktop\share」を変更
- 2016年08月03日講義内容について
- 予習事項(推奨)
- 2016年08月01日分の講義資料PDF(2016.07.31版; 約17MB)
- 2016年08月02日分の講義資料PDF(2016.07.31版; 約10MB)
- 2016年08月03日分の講義資料PDF(2016.07.06版; 約10MB)
- 2016年08月04日分の講義資料PDF(2016.08.12版; 約10MB)
- 予習事項(ほぼ任意)
- アグリバイオ大学院講義科目「農学生命情報科学特論II (特論II)」受講者用
予定ではDDBJ Pipeline実行部分(VelvetとPlatanus)は「エアーハンズオン」でしたが、
エアー(やったふり)だと当日時間を取る意義を見出せないので大幅に省略します。
DDBJ Pipeline実行結果ファイルの解析部分にできるだけ時間を費やしたいので、
第6回ウェブ資料PDF(2016.06.17版; 約20MB)のDDBJ Pipeline関連部分、
具体的にはW13からW17まで(スライド157から227まで)、およびW19-1からW20-4(スライド241から260まで)を眺めておいてください。
参加予定の平成28年度講習会資料について、目を通しておきましょう。 講習会当日は、できるだけ効率的に進めるために講義資料の一部を省略予定です。省略予定部分は特に目を通しておいてください。 随時差し替えます。
de novoアセンブリの理論の話(スライド134-147あたり)は、当日省略予定。 この部分は、第1部初日(2016年07月20日)のk-mer出現頻度分布の解釈や、シークエンスエラー由来k-merの除去などと関連します。 W8(スライド159-168あたり)も状況次第で省くかもしれません。
スライド10までは自習。当日はスライド11から始める予定です。スライド122-133のPacBioのイントロ部分は、疲れたころに初耳状態で聞くと相当疲弊すると思われます。 ここも当日は復習程度の位置づけで聞けるよう全体像を理解し頭を整理しておきましょう。
スライド8までは自習。当日はスライド9から始める予定です。
Trinityのapt-get経由でのインストール(スライド131-138)、およびBridgerのインストール(スライド139-186)は、当日省略予定。
第3部で利用するovaファイルは、日本乳酸菌学会誌のNGS連載第4回終了時点のovaファイル(URL情報はNGSハンズオン講習会事務局あてにメールで問い合わせてください)をもとに、 下記手順で作成しました。第4回終了時点のovaファイルをもらっておいて、自分で下記手順通りに環境構築してもおそらく大丈夫だろうと思います(2016年07月04日現在、最終確認中)。 全部を一度にコピペしてもうまくいきません。一行づつコピペして確実にダウンロードと確認をしていくのが鉄則です。
#################### ### 20160801用 ### #################### # W1-1 cd ~/Documents/srp017156 rm -f hoge* rm -f JS* rm -rf result* rm -f *.bz2 # W2-3 cd ~/Downloads wget -c http://cs.wellesley.edu/~btjaden/Rockhopper/download/current/Rockhopper.jar # W7-4 (「sudo R」のあとにログインパスワードpass1409を打つ) sudo R source("http://bioconductor.org/biocLite.R") biocLite("QuasR") # W7-5 (Update all/some/none? [a/s/n]:のところでaと打ってリターン) q(save="no") # W9-1 cd ~/Desktop mkdir backup cd ~/Desktop/backup wget -c ftp://ftp.ensemblgenomes.org/pub/bacteria/release-22/fasta/bacteria_15_collection/lactobacillus_casei_12a/dna/Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa.gz # W10-1 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_1.R # W12-3 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_2.R # W13-1 wget -c ftp://ftp.ensemblgenomes.org/pub/bacteria/release-30/fasta/bacteria_15_collection/lactobacillus_casei_12a/dna/Lactobacillus_casei_12a.GCA_000309565.2.30.dna.toplevel.fa.gz wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_3.R # W2-3の予備 wget -c http://cs.wellesley.edu/~btjaden/Rockhopper/download/current/Rockhopper.jar #################### ### 20160802用 ### #################### cd ~/Desktop/backup # W14-2 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_4.txt # W14-3 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_5.R # W16-1 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_6.R # W17-2 cp ~/.zshrc ~/Desktop/backup/.zshrc_org # W18-1 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_7.txt # W18-2 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB5_8.R # W3-2 (ここから連載第6回ウェブ資料) cd ~/Documents mkdir DRR024501 cd DRR024501 wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/DRA002/DRA002643/DRX022186/DRR024501_1.fastq.bz2 -O - | bzip2 -dc | head -n 1200000 | gzip > DRR024501sub_1.fastq.gz wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/DRA002/DRA002643/DRX022186/DRR024501_2.fastq.bz2 -O - | bzip2 -dc | head -n 1200000 | gzip > DRR024501sub_2.fastq.gz # W8-1 cd ~/Desktop/backup/ wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB6_1.R # W9-2 cd ~/Downloads wget -c http://www.ebi.ac.uk/~zerbino/velvet/velvet_1.2.10.tgz cp velvet_1.2.10.tgz ~/Desktop/backup/ #################### ### 20160803用 ### #################### # W11-2 (第6回ウェブ資料作成当時のバージョンをダウンロードできるのでそれにしている) cd ~/Downloads wget -c http://kmergenie.bx.psu.edu/kmergenie-1.6982.tar.gz cp kmergenie-1.6982.tar.gz ~/Desktop/backup/ # W12-1 cd ~/Desktop/backup/ wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/fastaLengthFilter.py # W18-1 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/DRR024501/DDBJP_Velvet_k131/velvet.zip # W20-4 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/DRR024501/DDBJP_Platanus/platanusResult.zip # W3-1 (ここから連載第7回ウェブ資料) cd ~/Desktop/backup/ wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/DRA002/DRA002643/DRX022185/DRR054113.fastq.bz2 # W4-1 (SRA Toolkitをwget経由でインストールしたかったヒト向け; 約61MB) # W4-1 (ln -sのシンボリックリンクは一例です。sudoがあるので、ログインパスワードを聞かれたらpass1409。) # W4-1 (最後のrehashはリロードみたいなもの。) cd ~/Downloads wget -c http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.6.3/sratoolkit.2.6.3-ubuntu64.tar.gz tar -zxvf sratoolkit.2.6.3-ubuntu64.tar.gz cd sratoolkit.2.6.3-ubuntu64 sudo ln -s /home/iu/Downloads/sratoolkit.2.6.3-ubuntu64/bin/fastq-dump.2.6.3 /usr/local/bin/ rehash # W6-3 cd ~/Desktop/backup/ wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/DRR054113/DRR054113_163380_fastqc.html # W9-3 wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/DRR054113/result.zip #################### ### 20160804用 ### #################### # 事前準備 cd ~/Desktop/backup/ wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2016/3/QC.1.trimmed.fastq.gz wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2016/3/QC.2.trimmed.fastq.gz # Trinity (約166MB) cd ~/Downloads wget -c https://github.com/trinityrnaseq/trinityrnaseq/archive/v2.2.0.tar.gz # Trinity実行結果ファイル (約6MB) cd ~/Desktop/backup/ wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2016/3/Trinity1.fasta wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2016/3/Trinity2.fasta # Bridger (約11MB) cd ~/Downloads wget -c https://sourceforge.net/projects/rnaseqassembly/files/Bridger_r2014-12-01.tar.gz # Boost (約85MB) cd ~/Downloads wget -c https://sourceforge.net/projects/boost/files/boost/1.61.0/boost_1_61_0.tar.bz2 # TIGAR2 (約6MB) cd ~/Downloads wget -c https://github.com/nariai/tigar2/archive/master.zip # FPKM値を手計算 cd ~/Desktop/backup/ wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2016/3/test_out.txt
特論IIの課題(kadai_tokuronII.pdf)と仮想NGSデータ(kadai_20160720.fasta)を2016年7月14日に公開しました。
提出期限は8月30日です。
第3部 | NGS解析(中~上級) | Linux環境でのデータ解析:JavaやRの利用法(2016.08.01)
「書籍 | 日本乳酸菌学会誌 | 第5回アセンブル、マッピング、そしてQC」に沿って進めます。 第5回W13-2までの内容です。
- 講義資料PDF(2016.07.31版; 約17MB)
第3部 | NGS解析(中~上級) | Linux環境でのデータ解析:マッピング、トリミング、アセンブリ(2016.08.02)
前半は「書籍 | 日本乳酸菌学会誌 | 第5回アセンブル、マッピング、そしてQC」に、 後半は「書籍 | 日本乳酸菌学会誌 | 第6回ゲノムアセンブリ」に沿って進めます。 前半は第5回W14-1以降、後半は第6回W10-6までの内容です。
de novoアセンブリの理論の話(スライド134-147あたり)は、当日省略予定です。 が、第3部初日(2016年08月01日)の、k-mer出現頻度分布の解釈や、シークエンスエラー由来k-merの除去などとも関連しますので、 必要に応じて予習(または復習)しておいてください。W8(スライド159-168あたり)も状況次第で省くかもしれません。
- 講義資料PDF(2016.07.31版; 約10MB)
第3部 | NGS解析(中~上級) | クラウド環境との連携、ロングリードデータの解析(2016.08.03)
前半は「書籍 | 日本乳酸菌学会誌 | 第6回ゲノムアセンブリ」に、 後半は「書籍 | 日本乳酸菌学会誌 | 第7回ロングリードアセンブリ」に沿って進めます。 前半は第6回W11-1以降、後半は第7回の内容(一部)です。 講習会当日は、スライド10までは自習とし、スライド11から始める予定です。 スライド122-133のPacBioのイントロ部分は、疲れたころに初耳状態で聞くと相当疲弊すると思われます。 ここも当日は復習程度の位置づけで聞けるよう全体像を理解し頭を整理しておきましょう。
- 講義資料PDF(2016.07.06版; 約10MB)
第3部 | NGS解析(中~上級) | トランスクリプトームアセンブリ、発現量推定(2016.08.04)
スライド107 (Trinityのパスを通すところのミス)、スライド140 (Bridgerに必要なboostパッケージをapt-getでやるとサンプルデータの実行もうまくいく)あたりを修正しました。(2016.08.12追加)
- 講義資料PDF(2016.08.12版; 約12MB)
- おさらい1: paired-end Illumina HiSeqデータ(SRR616268)
- サンプルは、Lactobasillus casei 12A (第3回W21)。ゲノム配列既知
- Ensembl Bacteria (release 22)当時はコンティグレベル (第3回W11; 第1回の図2)
- Ensembl Bacteria (release 30)頃は染色体レベル (第5回W13)
- オリジナルのリード数やファイルサイズ(第3回W23とW24)
- forward側(SRR616268_1.fastq.bz2):リード長は107 bp。ファイルサイズは7,662,128,101 bytes (約7.2GB)。
- reverse側(SRR616268_2.fastq.bz2):リード長は93 bp。ファイルサイズは7,017,031,734 bytes (約6.6GB)。
- サブセット(最初の100万リード)の取得(第4回W6)
- forward側(SRR616268sub_1.fastq.gz):リード長は107 bp。ファイルサイズは76,659,501 bytes (約74MB)。
- reverse側(SRR616268sub_2.fastq.gz):リード長は93 bp。ファイルサイズは68,682,959 bytes (約66MB)。
- 場所:~/Documents/srp017156
- FaQCs実行(第5回W1; 2016.08.01)
- forward側(QC.1.trimmed.fastq.gz)。ファイルサイズは73,669,994 bytes (約71MB)。
- reverse側(QC.2.trimmed.fastq.gz)。ファイルサイズは65,909,777 bytes (約63MB)。
- 場所:~/Documents/srp017156/result2
- 事前準備
- FastQC
- FastQCの結果を眺める
- forward側:data1_fastqc.html
- reverse側:data2_fastqc.html
- 場所:~/Desktop/mac_shareだが、ホストOS(~/Desktop/shareフォルダ)上で眺める
- Rockhopper2おさらい
- Tips:diff
- 確認
- 情報抽出(grep)
- 情報抽出2(grep)
- トリミング(fastx-trimmer)
- fastx-trimmer -f -l
- 様々なトリム条件1
- 様々なトリム条件2
- 結果の全体像を把握
- 様々なトリム条件3
- 結果の全体像を把握2 (スライド68)
- 他の結果もチェック1 (スライド73)
- 他の結果もチェック2 (スライド74)
- ファイル保存して整形 (スライド75)
- ベストな条件でpaired-endアセンブリを実行 (スライド78)
- Trinity:Grabherr et al., Nat Biotechnol, 2011
- ダウンロードとインストール (スライド89)
- パスを通す (スライド106)
- 色々試しながら実行1 (スライド109)
- 色々試しながら実行2 (スライド119)
- 色々試しながら実行3 (スライド128)
- apt-getでインストール (スライド132)
- Bridger:Chang et al., Genome Biol., 2015 (スライド141)
- Boost (スライド147)
- Bridgerに戻って作業 (スライド167)
- TIGAR2: Nariai et al., BMC Genomics, 2014 (スライド190)
- 推奨パイプラインでTIGAR2を実行 (スライド201)
- FPKM値を手計算 (スライド226)
スライド8までは自習。当日はスライド9から始める予定です。
Trinityのapt-get経由でのインストール(スライド131-138)、およびBridgerのインストール(スライド139-186)は、当日省略予定。
下記のファイルサイズは環境によって異なります。目安です。
ファイル名:Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa.gz (第3回W14-1でgenome.faに名前を変更)
場所:~/Desktop/hoge
ファイル名:Lactobacillus_casei_12a.GCA_000309565.2.30.dna.toplevel.fa.gz
場所:~/Documents/genomes
DRAから取得したbzip2圧縮ファイル。2つとも行数は539,023,984行、リード数は134,755,996リード(約1.35億)。大きすぎるのでBio-Linux8上には存在しない。
gzip圧縮FASTQファイルで合計約140MB。第4回W6でサブセット抽出を行い、第4回W9-7とW12-3でgzip圧縮しています。
1,000,000リード -> 977,202リード (第5回W1-3)。リード長はばらばら。第5回W14-1でgzip圧縮しています。
~/Documents/srp017156上に20160804ディレクトリを作成し、そこにgzip圧縮後のFaQCs実行結果ファイルをコピーして解析を行う。
cd ~/Documents/srp017156 mkdir 20160804 cd 20160804 pwd #FaQCs実行結果ファイルのコピー cp ~/Documents/srp017156/result2/QC.[0-9].*.gz . #cp ~/Desktop/backup/QC.[0-9].*.gz . ls -l #ファイル名変更 mv QC.1.trimmed.fastq.gz data1.fq.gz mv QC.2.trimmed.fastq.gz data2.fq.gz ls -l
FastQCを実行し、結果を共有フォルダ(~/Desktop/mac_share)に出力する。 fastqc2コマンドの実体はFastQC (ver. 0.11.4)であり、第4回W9-5でパスを通しています。 --nogroupは、「長いリードの場合に10番目以降の塩基を一定幅でグループ化する(これがデフォルト)」 機能をオフにするオプション(第5回W19-1)。約30秒。
cd ~/Documents/srp017156/20160804
pwd
ls -l
fastqc2 -v
fastqc2 -q --nogroup data1.fq.gz --outdir=/home/iu/Desktop/mac_share
fastqc2 -q --nogroup data2.fq.gz --outdir=/home/iu/Desktop/mac_share
ls -l ~/Desktop/mac_share/data*
最大メモリを2GBにしてpaired-endで実行(第5回W5-2)。約1分。
cd ~/Documents/srp017156/20160804 pwd ls -l java -Xmx2000m Rockhopper data1.fq.gz%data2.fq.gz ls ls -l Rockhopper_Results #ファイルの行頭と行末の8行分を表示 head -n8 Rockhopper_Results/summary.txt tail -n8 Rockhopper_Results/summary.txt
Rockhopper実行時に画面上に出力されるものをresult.txtに保存してスッキリさせる。 比較する2つのファイルの差分を表示させるdiffコマンドを実行し、何も表示されない(全く同じ)ことを確認。
cd ~/Documents/srp017156/20160804 pwd ls -l java -Xmx2000m Rockhopper data1.fq.gz%data2.fq.gz > result.txt ls ls -l Rockhopper_Results diff result.txt Rockhopper_Results/summary.txt
Rockhopper2の実行がほぼ一瞬で終わった理由は、おそらく最初にintermediaryなどの以前に実行したログ情報を見に行って、 全く同じファイル名やパラメータのものがあれば、それを返すような仕様にしているのであろう。 念には念を入れて、一旦Rockhopper_Resultsディレクトリを削除してもう一度実行する。 それなりの実行時間がかかり、diff結果で何も出なければ安心。
cd ~/Documents/srp017156/20160804 pwd ls rm -rf Rockhopper_Results rm -f result.txt ls -l java -Xmx2000m Rockhopper data1.fq.gz%data2.fq.gz > result.txt ls ls -l Rockhopper_Results diff result.txt Rockhopper_Results/summary.txt
Rockhopper_Results/summary.txtと同じ中身のファイル(result.txt)から、 とりあえず配列数(transcripts数)情報を抽出する。ここでは、"number of assembled transcripts" という文字列を含む行を表示させている。どういう文字列で抽出するかはlessなどでresult.txtを眺めておいて試行錯誤するのが一般的。
cd ~/Documents/srp017156/20160804 pwd ls -l grep "number of assembled transcripts" result.txt
single-endでforward側(data1.fq.gz)のみ、reverse側(data2.fq.gz)のみのRockhopper2を実行し、配列数を表示。
cd ~/Documents/srp017156/20160804 pwd ls -l java -Xmx2000m Rockhopper data1.fq.gz > result_f.txt java -Xmx2000m Rockhopper data2.fq.gz > result_r.txt ls -l grep "number of assembled transcripts" result*.txt
第5回W16-2で利用したfastx-trimmerを利用して、(とりあえず第5回W16-2と同じく100塩基目以降をトリムしたいので) 99塩基目まで残すという指定で実行し、trim1.fq.gzというファイル名で保存。
cd ~/Documents/srp017156/20160804 pwd ls -l data1* gunzip -c data1.fq.gz | head -n 4 gunzip -c data1.fq.gz | fastx_trimmer -l 99 - | gzip > trim1.fq.gz gunzip -c trim1.fq.gz | head -n 4 fastx_trimmer -h
「-f 3」を追加したら、「-l 99」はどうなるのかを調査。 つまり、「-f 3」は3塩基目からという意味なので、最初の2塩基分がトリムされる。 この場合に「-l 97」としないといけないのかどうかを知りたい。
cd ~/Documents/srp017156/20160804 pwd ls -l data1* gunzip -c data1.fq.gz | head -n 4 gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 99 - | gzip > trim1.fq.gz gunzip -c trim1.fq.gz | head -n 4
2種類のトリム条件で作成したtrim1.fq.gzを入力としてRockhopper2を実行し、配列数を得る基本形。 この段階で出力ファイル名の形式も意識する。
cd ~/Documents/srp017156/20160804 pwd ls -l data1* #-f 1:残す最初(first)の塩基が1塩基目という意味。 #-l 107:残す最後(last)の塩基が107塩基目という意味。 #つまり、トリムしないという意味。 gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 107 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_01_f_107.txt grep "number of assembled transcripts" result_01_f_107.txt #-f 1:残す最初(first)の塩基が1塩基目という意味。 #-l 104:残す最後(last)の塩基が104塩基目という意味。 #例えば元が104塩基より長いリードは3'末端側を104 bp分までトリムするが、 #FaQCs実行で元々104 bp以下のリードはトリムしない。 gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 104 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_01_f_104.txt grep "number of assembled transcripts" result_01_f_104.txt
多くのトリム条件を一気に計算する。3'末端のみ3塩基刻みで95 bpまでと、 5'末端を3塩基目からスタート(2塩基トリム)して3'末端を3塩基刻みで107-95 bpまでの長さを残す、 計8通り分を作成し、一気にコピペ実行
### -f 1 and -l 101 ### gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 101 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_01_f_101.txt grep "number of assembled transcripts" result_01_f_101.txt ### -f 1 and -l 98 ### gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 98 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_01_f_098.txt grep "number of assembled transcripts" result_01_f_098.txt ### -f 1 and -l 95 ### gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 95 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_01_f_095.txt grep "number of assembled transcripts" result_01_f_095.txt ### -f 3 and -l 107 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 107 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_03_f_107.txt grep "number of assembled transcripts" result_03_f_107.txt ### -f 3 and -l 104 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 104 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_03_f_104.txt grep "number of assembled transcripts" result_03_f_104.txt ### -f 3 and -l 101 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 101 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_03_f_101.txt grep "number of assembled transcripts" result_03_f_101.txt ### -f 3 and -l 98 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 98 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_03_f_098.txt grep "number of assembled transcripts" result_03_f_098.txt ### -f 3 and -l 95 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 95 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_03_f_095.txt grep "number of assembled transcripts" result_03_f_095.txt
lsでファイル名をざっくり眺めて、様々な条件でトリムした結果ファイル群のみをうまく表現できる「ワイルドカード(ここでは*)」を考える。 もちろんファイル名を眺めるだけでどのような条件でトリムしたかが一目でわかるように意味を持たせるのも基本。
cd ~/Documents/srp017156/20160804
pwd
ls
grep "number of assembled transcripts" result_0*
気になったトリム条件を実行。
### -f 1 and -l 99 ### gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 99 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_01_f_099.txt grep "number of assembled transcripts" result_01_f_099.txt ### -f 1 and -l 97 ### gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 97 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_01_f_097.txt grep "number of assembled transcripts" result_01_f_097.txt ### -f 3 and -l 99 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 99 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_03_f_099.txt grep "number of assembled transcripts" result_03_f_099.txt ### -f 3 and -l 97 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 97 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_03_f_097.txt grep "number of assembled transcripts" result_03_f_097.txt ### -f 2 and -l 99 ### gunzip -c data1.fq.gz | fastx_trimmer -f 2 -l 99 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_02_f_099.txt grep "number of assembled transcripts" result_02_f_099.txt ### -f 2 and -l 98 ### gunzip -c data1.fq.gz | fastx_trimmer -f 2 -l 98 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_02_f_098.txt grep "number of assembled transcripts" result_02_f_098.txt ### -f 2 and -l 97 ### gunzip -c data1.fq.gz | fastx_trimmer -f 2 -l 97 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz > result_02_f_097.txt grep "number of assembled transcripts" result_02_f_097.txt pwd ls -lt result_0*
「ワイルドカード(ここでは*)」をうまく利用して、着目したい結果ファイルのみを表示させて、全体像をよりわかりやすく把握できるような小細工もする。
Perfectly aligned reads:。
cd ~/Documents/srp017156/20160804
pwd
ls result_0*
grep "number of assembled transcripts" result_0*9[0-9].txt
Perfectly aligned reads行の結果もチェック。
cd ~/Documents/srp017156/20160804
pwd
ls result_0*
grep "Perfectly aligned reads" result_0*9[0-9].txt
Total number of assembled bases行の結果もチェック。
cd ~/Documents/srp017156/20160804
pwd
ls result_0*
grep "number of assembled bases" result_0*9[0-9].txt
Linux上でざっと傾向を見ておいて、あとはホストOS上でエクセルで手作業で整形してまとめたりします。 共有フォルダ(~/Desktop/mac_share)上に結果をファイル保存 リアルは、cutコマンドなどを用いたりしてもう少し手作業の手間を省いたりします。 コマンドを複数行にわたって記述する場合は「バックスラッシュ」です(第4回W5-2やW9-8)。 Macで「バックスラッシュ」を出したい場合は、「Alt + \」で出るらしい。
cd ~/Documents/srp017156/20160804 pwd grep "number of assembled transcripts" result_0*9[0-9].txt \ > ~/Desktop/mac_share/hoge_1.txt grep "Perfectly aligned reads" result_0*9[0-9].txt \ > ~/Desktop/mac_share/hoge_2.txt grep "number of assembled bases" result_0*9[0-9].txt \ > ~/Desktop/mac_share/hoge_3.txt
reverse側は何もトリムをしていないdata2.fq.gzを与えてpaired-endでRockhopper2を実行。 アセンブリ結果ファイル(Rockhopper_Results/transcripts.txt)はどんどん上書きされるので、別名でコピーしている。
pwd ### -f 1 and -l 99 ### gunzip -c data1.fq.gz | fastx_trimmer -f 1 -l 99 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz%data2.fq.gz > result_01_f_099_r_.txt grep "number of assembled transcripts" result_01_f_099_r_.txt cp Rockhopper_Results/transcripts.txt Rockhopper_Results/transcripts_01_f_099_r_.txt ### -f 2 and -l 99 ### gunzip -c data1.fq.gz | fastx_trimmer -f 2 -l 99 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz%data2.fq.gz > result_02_f_099_r_.txt grep "number of assembled transcripts" result_02_f_099_r_.txt cp Rockhopper_Results/transcripts.txt Rockhopper_Results/transcripts_02_f_099_r_.txt ### -f 3 and -l 99 ### gunzip -c data1.fq.gz | fastx_trimmer -f 3 -l 99 - | gzip > trim1.fq.gz java -Xmx2000m Rockhopper trim1.fq.gz%data2.fq.gz > result_03_f_099_r_.txt grep "number of assembled transcripts" result_03_f_099_r_.txt cp Rockhopper_Results/transcripts.txt Rockhopper_Results/transcripts_03_f_099_r_.txt grep "number of assembled transcripts" *_r_.txt grep "Perfectly aligned reads" *_r_.txt grep "number of assembled bases" *_r_.txt
講習会ではTrinity (ver. 2.2.0)のtar.gzファイルを~/Downloadsにダウンロード済み。
cd ~/Downloads
pwd
#wget -c https://github.com/trinityrnaseq/trinityrnaseq/archive/v2.2.0.tar.gz
ls -l v2*
tar zxvf v2.2.0.tar.gz
cd trinityrnaseq-2.2.0
ls
more README
make
ls -lt
~/binへのパスは第6回W12-3 (2016.08.03のスライド56)で通した。 「cp Trinity ~/bin」は間違い。 「ln -s /home/iu/Downloads/trinityrnaseq-2.2.0/Trinity ~/bin」が正解です(2016.08.12追加)。
cd ~/Downloads/trinityrnaseq-2.2.0
pwd
ls
where Trinity
#cp Trinity ~/bin
ln -s /home/iu/Downloads/trinityrnaseq-2.2.0/Trinity ~/bin
where Trinity
とりあえずFaQCs実行直後の977,202リードからなるdata1.fq.gzとdata2.fq.gzのpaired-endを入力としてTrinityを実行。
cd ~/Documents/srp017156/20160804 pwd ls -l data* ### COMMON.pmというPerlモジュール部分でエラー ### Trinity --seqType fq --max_memory 2G --CPU 2 \ --left data1.fq.gz --right data2.fq.gz ### COMMON.pm問題は解決。が、Phase 1のJellyfishの所でフリーズ状態に... ### ~/Downloads/trinityrnaseq-2.2.0/Trinity \ --seqType fq --max_memory 2G --CPU 2 \ --left data1.fq.gz --right data2.fq.gz ### 「CTRL + C」で脱出し、計算途中だったTrinity関連のものを念のため削除 ### ls -lt | head rm -rf trinity_out_dir ls -l *readcount rm -f *readcount
Trinity実行結果ファイル(Trinity1.fasta; 約3MB)の転写物数(2,603個)に衝撃を受ける。
cd ~/Documents/srp017156/20160804 pwd ls -l data* ### なんとなくメモリ1Gにして実行したらうまくいった ### ~/Downloads/trinityrnaseq-2.2.0/Trinity \ --seqType fq --max_memory 1G --CPU 2 \ --left data1.fq.gz --right data2.fq.gz ### 確認 ### ls -lt | head cd trinity_out_dir pwd ls grep -c ">" Trinity.fasta grep -v ">" Trinity.fasta | wc cp Trinity.fasta ../Trinity1.fasta
Trinity実行結果ファイル(Trinity2.fasta; 約3MB)の転写物数(3,090個; 別のPCでやると3,089個でした)から、strand情報を考慮すると配列数が増える(たまたまかも)と学習する。
cd ~/Documents/srp017156/20160804 ### 計算途中じゃなくてもTrinity関連のものを予め削除しないといけないようだ ### ### これをやらずに実行すると、すぐに計算が終わるのでやらねばと学習する ### ls -lt | head rm -rf trinity_out_dir ls -l *readcount rm -f *readcount ### SRR616268の実験情報SRX204226の記述(Illumina Stranded Std PE)から ### ### orientation情報も入れたほうがよいと判断して--SS_lib_type FRを追加 ### ~/Downloads/trinityrnaseq-2.2.0/Trinity \ --seqType fq --max_memory 1G --CPU 2 --SS_lib_type FR \ --left data1.fq.gz --right data2.fq.gz ### 確認 ### ls -lt | head cd trinity_out_dir pwd ls grep -c ">" Trinity.fasta grep -v ">" Trinity.fasta | wc cp Trinity.fasta ../Trinity2.fasta
apt-cacheでtrinityのキーワードを含むソフトウェア名をリストアップ。 正式名称がtrinityrnaseqであることを確認し、sudo apt-get installを実行。
cd pwd where Trinity ~/Downloads/trinityrnaseq-2.2.0/Trinity --version apt-cache -n search trinity sudo apt-get install trinityrnaseq where Trinity Trinity --version /usr/bin/Trinity --version
講習会ではBridger_r2014-12-01.tar.gz (約11MB)を~/Downloadsにダウンロード済み。 wget時のデフォルトのURLは「https://sourceforge.net/projects/rnaseqassembly/files/Bridger_r2014-12-01.tar.gz/download」でしたが、 得られるファイル名がdownloadとなって気持ち悪かったので、 「https://sourceforge.net/projects/rnaseqassembly/files/Bridger_r2014-12-01.tar.gz」に変更した。
cd ~/Downloads
#wget -c https://sourceforge.net/projects/rnaseqassembly/files/Bridger_r2014-12-01.tar.gz
pwd
ls -l Bri*
tar zxvf Bridger_r2014-12-01.tar.gz
cd Bridger_r2014-12-01
ls
more INSTALL
less README
講習会ではboost_1_61_0.tar.bz2 (約85MB)を~/Downloadsにダウンロード済み。 wget時のデフォルトのURLは「hhttps://sourceforge.net/projects/boost/files/boost/1.61.0/boost_1_61_0.tar.bz2/download」でしたが、 得られるファイル名がdownloadとなって気持ち悪かったので、 「https://sourceforge.net/projects/boost/files/boost/1.61.0/boost_1_61_0.tar.bz2」に変更した。 ver. 1.61ではうまくいかなかったが、apt-get経由で1.55をインストールするとうまくいくという情報を当日受講生からいただきました。 具体的には「apt-cache -n search boost」でパッケージ名を同定し、 「sudo apt-get install libboost1.55-all-dev」とやればうまくインストールできます。 成功すれば次はスライド167からのBridgerに戻ればいいです。(2016.08.12追加)
cd ~/Downloads #wget -c https://sourceforge.net/projects/boost/files/boost/1.61.0/boost_1_61_0.tar.bz2 pwd ls -l boo* bzip2 -dc boost_1_61_0.tar.bz2 | tar xvf - cd boost_1_61_0 ls ./bootstrap.sh pwd ls -ld b* cd boost pwd ls -ld include lib ### ./b2 install ### cd .. pwd ./b2 install --prefix=/home/iu/Downloads/boost_1_61_0/boost ### includeとlibの確認 ### pwd ls -ld boost/include boost/lib ### LD_LIBRARY_PATH ### tail -n 5 ~/.zshrc echo 'export LD_LIBRARY_PATH=/home/iu/Downloads/boost_1_61_0/boost/lib:$LD_LIBRARY_PATH' >> ~/.zshrc tail -n 5 ~/.zshrc echo $LD_LIBRARY_PATH source ~/.zshrc echo $LD_LIBRARY_PATH
cd ~/Downloads/Bridger_r2014-12-01 ### Configure ### pwd ls ./configure --with-boost=/home/iu/Downloads/boost_1_61_0/boost ### Make ### make ### Assemble -hで確認 ### pwd ls cd src ./Assemble -h ### small dataで動作確認 ### cd ~/Downloads/Bridger_r2014-12-01 pwd ls cd sample_test/ ls more run_Me.sh ./run_Me.sh ### 原因の特定を試みる ### ls -lt | head ls -l bridger_out_dir more bridger_out_dir/Assemble.log
講習会ではmaster.zip (約6MB)を~/Downloadsにダウンロード済み。
cd ~/Downloads #wget -c https://github.com/nariai/tigar2/archive/master.zip pwd ls -l mas* ### 解凍 ### unzip master.zip ### Githubと照合 ### pwd head tigar2-master/README.md ### 動作確認 ### java -jar ~/Downloads/tigar2-master/Tigar2_1.jar
TIGAR2の、「Recommended pipeline to run TIGAR2」の部分です。
### 1. FASTA形式のリファレンス配列を準備 ### cd ~/Documents/srp017156/20160804 ls -la Trinity1.fasta #wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2016/3/Trinity1.fasta grep -c ">" Trinity1.fasta grep -v ">" Trinity1.fasta | wc ### 2. マッピングプログラムbowtie2用のインデックスを作成 ### bowtie2-build --version mkdir rof bowtie2-build Trinity1.fasta ./rof/trenitey ls -l rof ### 3. マッピングプログラムbowtie2を実行 ### ### トリム前のpaired-endでとりあえずやる ### bowtie2 --version bowtie2 -h bowtie2 -p 2 -k 100 --very-sensitive -x ./rof/trenitey \ -1 data1.fq.gz -2 data2.fq.gz > test.sam pwd ls -l test.sam ls -lh test.sam ### 4. TIGAR2を実行 ### pwd ls -l Trinity1.fasta test.sam java -jar ~/Downloads/tigar2-master/Tigar2_1.jar \ --thread_num 2 Trinity1.fasta test.sam \ --is_paired --alpha_zero 0.1 test_out.txt ls -l test* head -n 5 test_out.txt grep ">" Trinity1.fasta | head -n 5
TIGAR2実行結果ファイル( test_out.txt の4列目のFPKM値を、2列目の配列長情報(LENGTH)および3列目のマップされたフラグメント数情報(Z)を用いて手計算する。 oeの部分は任意の文字で構いません。 $3は3列目という意味です。
cd ~/Documents/srp017156/20160804 #wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2016/3/test_out.txt #cp ~/Desktop/backup/test_out.txt . cat test_out.txt | awk '{oe=oe+$3} END{print oe;}' head -n 5 test_out.txt ### Rを起動してFPKM値を手計算 ### R -q 11 * (1000000/971222) * (1000/643) # "TRINITY_DN1285_c0_g1_i1" 56 * (1000000/971222) * (1000/325) # "TRINITY_DN1238_c0_g1_i1" 8 * (1000000/971222) * (1000/303) # "TRINITY_DN1264_c0_g1_i1" q(save="no")
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2015
NGSハンズオン講習会を2015年7月22日-8月6日の11日間(A日程)、 および予備日の8月26-28日(B日程)で実施しました。アメリエフ様分(服部先生と山口先生)の講義資料を差し替えました(2015/09/03)。 講義映像・動画が統合TVおよびyoutubeから公開されました(2015/11/13)。
はじめに(全員目を通しておきましょう)
- 講習会期間中アグリバイオノートPCを借りるヒトは、7/22の動作確認作業自体は行う必要は基本的にありません。
しかし、7/22のところに列挙した項目の予習は必須です。「エアーハンズオン」をするなりしてチェックリスト項目を
クリアしておきましょう。
- 平成26年度開催のNGS速習コース関係
- 報告書PDF(h26_ngs_report.pdf; 約4MB)
- 報告プレゼン資料(20150126_kadota.pdf; 約1MB)
- 平成27年度開催のNGSハンズオン講習会関係
- 前座プレゼン資料(20150722_kadota.pdf; 2014.07.14版; 約1MB)
- ovaファイルを用いたBioLinux8インストール法(20150722_BioLinux8_ova_win.pdf; 2014.07.22, 12:18版; 約1MB)
- 8月3-5日受講予定者は、この期間のどこかで利用予定の コマンドファイル(command.zip)をダウンロードしておいてください。(2015/07/28)
- Macのテキストエディタに関する注意点(野間口達洋 氏提供情報)。(2015/07/29)
- 受講者アンケート
概要、スケジュール、アンケート結果、受講生のコメントなどを見られます。
報告書PDFの短縮版のようなものです。Twitterやっているヒトは、ハッシュタグ#AJACS
をつけて平成27年度も有効利用してください。
概要、注意事項、受講生の心構えなどをざっくりと書いてあります。
7/22の環境構築済みのovaファイルを用いた簡単にインストールできるやり方です。自身のPCで環境構築に失敗、あるいは変なことになっちゃっても、 10数分程度で7/22終了時までの状態に復旧できます。HDD容量の事情にも合わせ150GB版と50GB版の2種類を用意しています。 また、7/23終了時点のものも講習会会場にUSBで配布しております。Macでは動作確認できていませんが、おそらく大丈夫だと思います。 isoファイルからのインストールよりも圧倒的に楽です。安心して沢山失敗しまくってくださいw
中身はReseq_command.txtと RNA-seq_command.txtです。
アンケート回答期間は平成27年7月22日(水)~9月11日(金)。 後日、メールにてご連絡いたします認証ID. およびPWを入力してご回答下さい。
2015年7月22日(2015.07.22):Bio-Linux 8とRのインストール状況確認(門田幸二、寺田 透)
書籍 | 日本乳酸菌学会誌 | についてで示した通りのPC環境を構築しておきましょう。 連載第1-3回、および第4回のウェブ資料W6-5までの予習は必須です。Rについても同様です。 一週間程度はきっちり時間をかけて予習しておきましょう。 7/22は、以下に示すようなことができる(わかる)ようになっていることの確認を自分でしてもらう日です。 門田自身全てを完全に把握しているわけではありませんし、ウェブ資料のページ数も膨大ですので、 どこにどういうことが書かれていたかの全体像の俯瞰やチェックリストという位置づけでもあります:
- Bio-Linux 8
- (第4回だけでなく第2-3回のウェブ資料PDFにも目を通している)
- ゲストOS、ホストOSなどと言われてうろたえない(第1回)
- Linux環境では、一般にダブルクリックでソフトウェアのインストールはしない(第1回)
- Perl, Ruby, Pythonはプログラミング言語である(第1回)
- 「円(yen)マーク」と「バックスラッシュ」の違いは見栄えの問題であり、文字コードとしては同じなので気にしない(第2回)
- コマンドとオプションの間には「スペース」が入るものだ(第2回)
- .(どっと)から始まるファイル名のもの(の多く)は、環境設定ファイルである(第2回)
- 全角文字をコマンドとして打ち込むのは非常識である(第2回)
- ファイルの拡張子は常識的に使う。例えばFASTQ形式ファイルの拡張子を.fastaや.faなどにしない(第2回)
- 「Tabキーによる補完」、「上下左右の矢印キー」を駆使して効率的にコマンドを打てるようになっている(第2回)
- リダイレクトやパイプを含むコマンドを眺めて、何をやっているものかが大体わかるようになっている(第2回)
- Bio-Linuxの左側の引き出しみたいなアイコンまたはターミナル上で、 ホスト <--> ゲスト間のファイルのやり取りが自在にできる(第3回)
- ホストからゲストへはドラッグ&ドロップ可能だが、ゲストからホストへはドラッグ&ドロップ不可能[W8-1](第3回)
- ターミナルの画面サイズや不具合は自分で変更および対処可能[W9-2-4](第3回)
- 共有フォルダの概念を理解している[W9-3](第3回)
- rmコマンドは「ゴミ箱」への移動ではなく本当に消える[W10-4](第3回)
- ダウンロードしたいファイルのURL情報取得ができる[W11-1](第3回)
- ゲストOSのネットワーク接続が不調の場合は、つながるまで待つのではなく、ホストOS上で必要なファイルをダウンロードして共有フォルダ 経由でゲストOS上の適切な場所に置くことができる[W11-1](第3回)
- 圧縮・解凍コマンド(zip, unzip, gzip, gunzip, bzip2, and bunzip2)の使い方をマスターしている[W13](第3回)
- headやtailコマンドでファイル中の最初と最後の部分を確認することができる[W14](第3回)
- cat, more, lessコマンドの基本的な使用法(脱出したいときはCTRL + Cなど)を理解している[W14-4からW14-6](第3回)
- 多くのLinuxコマンドには様々なオプションがあり、--hや--helpなどで利用可能なオプションを眺めることができる[W15-2](第3回)
- PDFファイルやPowerPointファイル中のコマンドをコピペするときは気をつける[W16-1](第3回)
- コマンドオプション利用時の「ハイフン」と「ダッシュ」の気をつける[W16-3](第3回)
- 初心者向けテキストエディタgeditの基本的な利用法は知っている[W17](第3回)
- 仮想環境全体のディスク使用状況把握(dfコマンド)や、カレントディレクトリのディスク使用量(duコマンド)を把握し、 適切に不要なファイルを削除できる[W18-2からW18-4](第3回)
- 「.(どっと)」はカレントディレクトリを表し、「..(どっとどっと)」は1つ上の階層のディレクトリを意味する[W18-3](第3回)
- 複数のコマンドを組み合わせたパイプ(|)利用の基本を知っている[W19](第3回)
- 連載原稿中では乳酸菌RNA-seqデータ(SRR616268)を例に解説しているが、 NGSハンズオン講習会(または予習)では、(自己責任で)自分が興味あるデータで平行して解析してもよいことを知っている[W20以降](第3回)
- bzip2はgzipに比べて圧縮率は高く、圧縮に要する時間も早いが、解凍は遅い[W22-5からW22-6](第3回)
- ファイルサイズが大きいものは、一つ一つの処理に要する時間がとてつもなく長くなることを知っている[W23](第3回)
- コマンドオプション、パイプ、リダイレクトを駆使することでNGSデータのサブセットを抽出できることを知っている[W25-2](第3回)
- アスタリスク(*)などのワイルドカードというものがあり、それらの挙動がある程度わかる[W1-2からW1-3](第4回)
- ゲストOSがフリーズ状態に遭遇しても、自力で再起動するなりして復旧できる[W2-1](第4回)
- Permission問題はchmodで自力で解決できる[W3-1](第4回)
- 同じ目的を達成する上で様々なやり方があることを知っている[W3-2](第4回)
- ファイルのダウンロード時に拡張子がオリジナルと変わっていることがある[W4-4](第4回)
- 自動マウント(オートマウント)の設定ができている[W4-5; W4-7](第4回)
- 絶対パス、相対パスの概念が理解できている[W4-6](第4回)
- 文字コード(改行コード)をodやfileコマンドで確認し、perlで変換できる[W5-2; W5-3](第4回)
- R
- インストール | についてをよく読み、ここに書いてある手順に従って2015年4月4日以降にインストールを行った
- インストール | についてで書いてある内容はBio-Linux8(ゲストOS)とは無関係であり、 WindowsやMacintosh(つまりホストOS)上で行う作業である
- ファイルの拡張子(.txtや.docxなど)はちゃんと表示されている
- Rの起動と終了ができる。終了時に表示されるメッセージにうろたえない
- R本体だけでなくRパッケージ群のインストールもちゃんと行った
- Rパッケージ群のインストール確認も行い、エラーが出ないことを確認した
- library関数を用いたRパッケージのロード中に、別のパッケージがないことに起因するエラーメッセージが出ることもあるが、 必要なパッケージを個別にインストールするやり方を知っている
- 基本的な利用法をよく読み、予習を行った
- 作業ディレクトリの変更ができる
- 例題ファイルのダウンロード時に、拡張子が勝手に変わることがあるので注意する
- 慣れないうちは、getwd()とlist.files()を打ち込むことで、作業ディレクトリと入力ファイルの存在確認を行う
- エラーに遭遇した際、「ありがちなミス1-4」に当てはまっていないかどうか自分で確認
2015年7月23日(2015.07.23):Linux基礎(門田幸二)
講義資料 (2015.07.21版; 約10MB)
- Bio-Linux:Field et al., Nat Biotechnol., 2006
- テストデータ取得:amelieff.zip (約39MB; 40,445,616 bytes; 2015.07.16更新)
- IGV(Thorvaldsdóttir et al., Brief. Bioinform., 2013)のインストール
- FastQC:原著論文なし
- GATK(ver. 1.6-13; McKenna et al., Genome Res., 2010)
wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/bioinfo_ngs_sokushu_2015/amelieff.zip
wget -c http://data.broadinstitute.org/igv/projects/downloads/IGV_2.3.57.zip
JSLAB4_2_Linux2.sh (Linuxの改行コード; LF)[W7-8のちょっと改変版]
wget -c http://www.iu.a.u-tokyo.ac.jp/~kadota/book/JSLAB4_2_Linux2.sh 中身は以下のもの: #wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061483/SRX204226/SRR616268_1.fastq.bz2 #wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061483/SRX204226/SRR616268_2.fastq.bz2 #bzip2 -dc SRR616268_1.fastq.bz2 | head -n 4000000 | gzip > SRR616268sub_1.fastq.gz #bzip2 -dc SRR616268_2.fastq.bz2 | head -n 4000000 | gzip > SRR616268sub_2.fastq.gz fastqc -q SRR616268sub_1.fastq.gz fastqc -q SRR616268sub_2.fastq.gz
sudo cp GenomeAnalysisTK-1.6-13-g91f02df.tar.bz2 /usr/local/src sudo chmod 755 GenomeAnalysisTK-1.6-13-g91f02df.tar.bz2 sudo bzip2 -dc GenomeAnalysisTK-1.6-13-g91f02df.tar.bz2 | sudo tar xvf -
2015年7月24日(2015.07.24):シェルスクリプト(アメリエフ株式会社 服部恵美先生)
講義資料 (約2MB; 2015.09.02版)
-
【内容】シェルスクリプトのメリットは、①効率的に解析できること、②バイオインフォは日進月歩だがLinuxコマンドやシェルスクリプトの文法は
大きく変わっていないので一度身につければ長く使える。シェルとは、ユーザが入力したコマンドをコンピュータに伝えるプログラムであり、bash、
zshなどいくつかの種類がある。講習会では、Bio-Linuxのデフォルトであるzshをベースとした記述になっている。シェルスクリプトの作成と実行。
viやgeditなどのテキストエディタで実行内容をファイルに書いて保存したのち、shコマンドで実行。主に使ったのは、geditのほう。
テストデータは、FASTA形式ファイル。到達目標は、「FASTAファイルから指定した遺伝子の配列だけを取り出す」。
最初は「head –n 4 ファイル名」のような簡単な記述内容のファイル(gedit test1.sh)からスタートし、sh test1.shで実行。
最初の8行分を表示するには?などの問いから徐々に難易度を上げていった。同じファイル名のものを何度も書くのは間違いのもとであり、
面倒なので、変数を使って値を一元管理するやり方。「変数名=値」と書いたのち、$変数名として変数に入っている値を呼び出すことができる。
スクリプト内に何度も登場するゲノム配列のファイル名などは変数にしておいたほうが便利。対象ファイルが変わるたびにスクリプトファイル内
のファイル名の値を変更しないといけない。その場合は、「引数」を使って実行するファイル名を外から与える。
引数は、実行時にスクリプト名以降に入力された値(空白区切りで複数入力可)。引数の値は、専用の変数に入る。変数$1に1番目の値、$2に2番目の値、など。
条件付き処理や、繰り返し処理、標準出力と標準エラー出力。シバン、cdbtools、最終課題へと進んだ。
2015年7月27日(2015.07.27):Perl(アメリエフ株式会社 服部恵美先生)
講義資料 (約2MB; 2015.09.02版)
-
【内容】Perlは、ファイル形式の変換などに利用される。高速な処理には向かないが、比較的手軽に書けてテキスト処理が得意なことから、
バイオインフォマティクス業界ではよく使われている。Perlはゆるい。同じ処理を様々な書き方で書ける言語である。
シェルスクリプトとの主な違いは、Perlのほうが、正規表現・複雑な計算や処理に向いている。ファイル形式の変換(BAM BED)や、
異なるプログラムの出力結果をマージするなど独自解析ができるのがPerlのいいところ。到達目標は、FASTAファイルのID行を変更したり、
アミノ酸の出現頻度を数えたり…。
Perlの文法。printを例として、行の末尾に;をつけるとか、全角記号・全角空白は使ってはいけないなど。シェルスクリプトのechoと違って、
改行する場合には明示的に\nを書く必要がある。Perlでも変数を使うことができる。基本形は「my $変数名=値;」であり「$変数」と書くと、
変数に入っている値を呼び出すことができる。myに関連して、スコープの話。値が沢山あるときは、「配列」を使ってまとめて取り扱うことができる。
配列とは、複数の値を1つの名前でまとめたもの。「my @配列名=(値,値,…);」のように書く。値を取り出すときの「添字」や、添字が0から始まることなど。
pushを使った値の追加、配列の要素数。配列を結合して文字列にする(join)、文字列を分割して配列にする(split)。コマンドライン引数@ARGVの説明。
値が沢山あって各データに名前を付けたいときは、「ハッシュ」を使えばよい。ハッシュへのキーと値の追加。Perlでも条件つき処理や繰り返し処理が可能。
Perlの比較演算子。whileやforの具体例。繰り返し処理中での次の処理へのスキップ(next)や処理の中止(last)。{}で囲んだ範囲をスコープと呼ぶ。
スコープ内で定義した変数は、そのスコープ内でのみ有効(ローカル変数と呼ぶ)。ファイル入出力、コメント、シバン、正規表現、メタ文字。
2015年7月28日(2015.07.28):Python(アメリエフ株式会社 服部恵美先生)
講義資料 (約2MB; 2015.09.03版)
- ヒトに渡すスクリプトはPython、自分しか使わない or 1回しか使わないスクリプトはPerlがいいのではないだろうか。
- Python Documentation contents
- Biopython
- codeacademy
- UをTに変換するような場合に、小文字を入力しても認識してくれるようにしたいときは、.upper()や.lower()をつけるとよい。
- Python3だとBioLinux8にBioモジュールがデフォルトでインストールされていない。
- vi講座。コマンドモードで:を押して「set nu」で左側に行番号をつけることができる。
- undo, redoは「u」「CTRL + r」。CTRL + uでアップ、CTRL + dでダウン。 ^は行の最初、$は行の最後(孫建強氏 提供情報)。 :w test2.txtでファイル名を指定して保存。上書き保存の場合は、:wのみでよい。 :wqが保存して終了。ZZでも上書き保存可能。:q!で保存せずに強制終了できる。 CTRL + vで矩形選択モード。:%s/nuc/aa/gcで、一つ一つ見ながらy or nで置換していくことができる。 コマンドモードでvでビジュアルモード。これで任意の範囲を選択できる。好きな場所でpを押す。 :%s/$/;で行末に一括で;を挿入。:set encoding=Shift-JISでエンコーディングを設定。:set enc?で確認。 :sav test4で保存した後、それを開くことができる。
2015年7月29日(2015.07.29):R基礎(東大 門田幸二)
講義資料 (2015.07.27版; スライド136枚でないヒトはリロード; 約6.7MB)
2015年7月30日(2015.07.30):Bioconductorの利用法(東大 門田幸二)
講義資料 (2015.07.31版; 約7MB)
- ??alphabetFrequency実行結果は、MacとWin間で異なる。
- available.genomes()実行結果などで特定のキーワード(ex. 'Hsap')を含む行のみを抽出して表示させたい場合は、 grep('Hsap', available.genomes())などとすればよい。(甲斐政親氏 提供情報)
- イントロ | 一般 | 配列取得 | プロモーター配列 | GenomicFeatures(Lawrence_2013)で、 取得したい領域がsense側のみゲノム配列の範囲内に収まるものを抽出する条件判定はしているものの、antisense側の条件判定は未実装であり改善が必要(2015/7/30の講義終了時点)。 -->取得したい領域がsense, antisense両方ともゲノム配列の範囲内に収まるものだけを抽出して出力するよりよいコードになった(甲斐政親氏 提供情報; 2015/08/03追加)。
genome <- available.genomes()
grep('Hsap', genome)
hit <- grep('Hsap', genome)
genome[Hit]
2015年8月3日(2015.08.03):NGS解析。基礎(アメリエフ株式会社 山口昌雄先生)
講義資料 (約3MB; 2015.09.02版)
- command.zip
- samtools viewだけで打つと、ヘルプがでる。[options]の[]は、あってもなくてもいい。 <in.bam>|<in.sam>で、パイプ(l)は、どちらが入力でもいいという意味。 <>は入力として絶対必要なもの。
- samファイルはマップされなかったリードも出力する
- 「samtools ngs」などで検索してsamtoolsのサイトにいく。 右上のほうにマニュアルがある。1列目の情報などの詳細を見ることができる。ときどき改訂されているようだ。
- CIGAR記号の説明。12MのMは一致、4IのIはinsertion、など。
- vcfファイルの説明。
- FASTA形式は、NCBIは70文字で改行を入れる。lessとgrepを利用。
- DDBJ SRA (DRA)でERR038793を検索。
- 午後は、igvから。less igv.shでファイルの中身を表示。 デフォルトは-Xmx2000mになっている。デカいファイルの場合は、この2000を4000などに変えて利用する。
- Windowsのヒトは、igv.batをダブルクリックで起動すればよい。これは32 bit版のJavaが1.2GBが上限だったという理由がある。
- 「samtools view 1K_ERR038793.bam | less」とすればsamに変換した結果をそのままlessで見ることに相当。
- samファイルのデフォルトは、リードのIDの並びになっている。これを染色体の並び順にソートするのが、「samtools sort」。 「samtools sort」だけでリターンを打つと、ヘルプが出る。<out.prefix>は、ファイルの拡張子なしの部分。
- 「samtools sort」の前に「samtools index」をやっても怒られる。samtools indexの結果として、.bam.baiができる。 このbaiは、bamのindex、に由来するのだろう。
- 「File - Load from File」でbamファイルを選ぶ。*_sort.bamを選ぶ。ソートされてないものを選ぶと怒られる。
- GUI左下の「Gene」のところで右クリックすると、collapsedやSquishedなどいろいろ選べる。
- bamがあって、baiファイル作成を忘れても、「Tools」メニューの「Run igvtools」で作れるのでべ便利。
- 「Genomes」メニューの「Create.genome File」でゲノムファイル以外にGene fileなどでおそらくGFF3などアノテーションファイルも読み込ませることができる。
- 「firefox fastac_report.html」でFireFoxが起動してhtmlファイルを見ることができる。
- NGS機器の原理の理解はyoutubeなどがいいかも。
- Nagasaki et al., Nat Commun., 2015
- velvetのインストール。普通は、/usr/local/srcなどにソースファイルを置く。
- マッピングで、segmental duplication (せぐでゅぷ、とも略すらしい)やリピート領域を除外する戦略もあり。 しかしsegmental duplication領域に重要な遺伝子が見つかったりすることもあるので結構ビミョー。
- 「ngs best practice」が出始めている。
- bwaやtophat-fusion
- 一つの研究プロジェクトなど、再現性が重要な場合があるときは、ず~っと同じプログラムのバージョンにする。 致命的なバグの場合は、もちろんバージョンを上げる。
- topコマンドでメモリ使用量を見る。SEQanswers
- velvetをアドリブで実行。 以下を実行するとtmpディレクトリが作成されて、その(tmpディレクトリ)中にcontigs.faというアセンブル結果ファイルができる。
- velvetをGUIベースで簡単にやるソフトウェア Vague(Powell and Seemann , Bioinformatics, 2013) をインストールして実行。
cd ~/Downloads
wget -c http://www.ebi.ac.uk/~zerbino/velvet/velvet_1.2.10.tgz
cd velvet_1.2.10
make 'MAXKMERLENGTH=79'
cd ~/Desktop/amelieff # single-endでやる場合 velveth tmp 31 -fastq 1K_ERR038793_1.fastq velvetg tmp grep -c ">" tmp/contigs.fa # paired-endでやる場合 velveth tmp2 31 -shortPaired -separate -fastq 1K_ERR038793_1.fastq 1K_ERR038793_2.fastq velvetg tmp2 grep -c ">" tmp2/contigs.fa
cd ~/Downloads wget -c http://www.vicbioinformatics.com/vague-1.0.5.tar.gz tar zxf vague-1.0.5.tar.gz cd vague-1.0.5 ./vague
2015年8月4日(2015.08.04):NGS解析。ゲノムReseq、変異解析(アメリエフ株式会社 山口昌雄先生)
講義資料 (約3MB; 2015.09.02版)
- QC用プログラムは、FASTX-toolkit(原著論文はない)やTrimmomatic (Bolger et al., Bioinformatics, 2014)などが最近よく使われる。
- Reseqはステップごとにbamファイルがどんどん作成されていくので、FASTQファイルサイズの5倍程度のHDD容量を確保しておいたほうが無難。
- GATKの後のアノテーションは、フリーソフトのsnpEff (Cingolani et al., Fly (Austin), 2012)が最近よく使われる。
- ゲノムファイルの取得は、Illuminaのigenomeも結構使われるので便利。 ヒトデータに限って言えば、NCBIとUCSCはかなり似ている。Ensemblはちょっと毛色が違う。hg38はNCBIとUCSCは統一された。RNAはEnsemblが力を入れている印象。
- 解凍は、tar zxvf *.tar.gzのオプションが基本。vオプションはあってもなくてもよい。
- Ensembl, RefSeq, UCSCでどのアノテーションを使うのがいいかに関する評価研究論文。Zhao and Zhang, BMC Genomics, 2015
- GRCh38はデコイ配列を含む。但し、ゲノム解析系はアノテーション情報がついてこれてないので移行しづらい。 GRCh38は、1000人ゲノムプロジェクトのコンセンサスを元に作成。
- スライド15はリンクのlなどと書いてあるが、実体のファイルがある。
- スライド17で「ln -s ../../WholeGenomeFasta/genome.fa」と書いているが、これは省略形。 「ln -s ../../WholeGenomeFasta/genome.fa ./genome.fa」が正式。 同じファイル名の場合は省略してよくて、カレントディレクトリ上に作成する場合も省略してよい。
- headとtail両方で確認。tailで確認するのは、コピー時におしりの部分が欠けることがあるので、それが分かる。
- fastq_quality_filter (ver. 0.0.14)コマンドで「-Q 33」はつけなくても大丈夫そう。
- スライド32で、「bwa mem 2> hoge」とすることでマニュアルをhogeに書きだしてくれる。
- samtools viewのデフォルトは、bamをsamにするが、-Sを指定するとinputがsamであることを意味し、 -bを指定するとoutputがbamであることを意味する。
- 実用上は、パイプを駆使して無駄に多くのファイルを作成しない。「-」はパイプでわたってきた入力ファイルの意味。 「samtools sort」はメモリを食うのでsortだけやらないこともある。ちなみにこの次のsamtools indexは独立してやる。 理由はこれまでパイプでつなぐと、bamファイルがない状態でbamのindexファイル(*.bam.bai)だけができてしまうことになるから。
- スライド37で3列目のマップされたリードをカウントする場合は$3、4列目のマップされなかったリード数は$4にすればよい。
- おまけ?!tviewオプションでIGVのような簡単なマッピング結果を眺めることもできる。
- bwaのマッピング結果は必ずしも信頼できるわけではないので、 GATKを使って怪しい領域を抜き出して丁寧にアラインメントをやり直す必要がる。これをre-alignmentという。
- スライド38。「-glm」がgenotypingのアルゴリズム。-glmをいれなくてもデフォルトのSNPはやってくれる。 -glm BOTHでSNPとINDELの両方を同時にやってくれる。別々にやって別ファイルに書きだすのもアリ。
- GATK実行時にエラーが見える(Error Response)が、こういう場合はGATK Support Forum。
- dbSNPに登録されている場合はそれを利用してよりよい結果を返すこともできる。 また複数サンプルを同時に入力として与えて検出の信頼度を上げることもできるらしいが、 アメリエフさんはまだやったことがないらしい。
- スライド41は94個の変異となったが、実際にやると100個となった。
- IGVで読み込んで、vcfファイルの水色がhomozygoteで青がheterozygote
- スライド47。「GATK VariantFiltration」で検索。
- vcftools
bwa mem -R "@RG\tID:1K_ERR038793_1\tSM:ERR038793\tPL:Illumina" /home/iu/Desktop/amelieff/Scerevisiae/BWAIndex/genome.fa 1K_ERR038793_1_qual.fastq | samtools view -Sb - | samtools sort - hoge_sorted
samtools idxstats 1K_ERR038793_1_qual_sorted.bam > tmp
awk '{a += $3} END {print a}' tmp
samtools tview 1K_ERR038793_1_qual_sorted.bam
2015年8月5日(2015.08.05):NGS解析。RNA-seq前半(アメリエフ株式会社 山口昌雄先生)
講義資料 (約4MB; 2015.09.03版)
- 画面の拡大または縮小は、「CTRLと+」または「CTRLと-」
- 融合遺伝子検出は難しい。deFuseやTopHat-Fusionなどがある。
- DDBJ SRA (DRA)の見方概要。
- fastqcコマンド実行時に「-t 2」オプションをつけると、CPU2個を使って処理するので早くなる。
- fastx_trimmerのxは、FASTQ/FASTAどちらでも対応可能という意味。
- スライド20は、最初の1塩基と書いている。が、どう考えても最初の7塩基をトリムしたほうがよさそうなのでそうする。
- スライド21で、8塩基目から使いたいので、とりあえず以下のように実習では打った。「-l 82」は、82塩基目までを残して、それ以外はトリムするというもの。
- スライド22で、fastx_clipperのオプション。-lはデフォルトが5だが、マッピングプログラムの性能的に20塩基程度はないとマップできないので、-l 20とする。
- スライド23で、prinseqを推奨。リード中のPolyA/Tの長さに合わせて自動的にトリムしてくれる。
- tagcleanerは捨てすぎる。fastx_clipperはあまり捨ててくれない。cutadaptはちょうどよかった。
- fastq_quality_trimmerで、順番は大事。最初にquality20未満をトリムしてから、80%以上がquality20以上などを残す。
- TopHatのマニュアルを眺める。
- スライド29で、-oで作成するディレクトリ名、-Nが許容するミスマッチ数(デフォルトが2)、--read-edit-distがedit distance。これはbest practiceによく書かれていたから。 「....Bowtie2Index/genome」までしか記述せず、拡張子は書かないのがポイント。
- スライド32
- スライド33で、~/Desktop/amelieff/rnaseq/1K_SRR518891中のjunction.bedもIGVで読み込ませた。「II:45,600-46,000」あたりのジャンクションリードを眺めた。
- スライド36
- igenomesに情報がある場合は、それを使うのが王道。UCSCのTable Browserで各生物種のgenes.gtfまでその場で作成できる。アノテーションファイル取得に関するTips。ただし、遺伝子名の列が間違ったりして修正が大変。
cd rnaseq fastx_trimmer -f 8 -l 82 -i ../1K_SRR518891_1.fastq -o 1K_SRR518891_1_s.fastq
fastx_clipper -a AAAAA -i 1K_SRR518891_1_s.fastq -o 1K_SRR518891_1_s_notail.fastq
fastq_quality_trimmer -t 20 -l 30 -i 1K_SRR518891_1_s_notail.fastq | fastq_quality_filter -q 20 -p 80 -Q 33 -o 1K_SRR518891_1_clean.fastq
tophat -o 1K_SRR518891 -g 3 -N 10 --read-edit-dist 10 --read-gap-length 10 /home/iu/Desktop/amelieff/Scerevisiae/Bowtie2Index/genome 1K_SRR518891_1_clean.fastq
ls 1K_SRR518891
samtools index 1K_SRR518891/accepted_hits.bam igv.sh
cufflinks --min-frags-per-transfrag 2 -o 1K_SRR518891 1K_SRR518891/accepted_hits.bam ll -h 1K_SRR518891
2015年8月5日(2015.08.05):NGS解析。RNA-seq後半(東大 門田幸二)
講義資料 (約3MB)
-
【内容】カウントデータ取得以降の統計解析。7/29のR基礎で簡単に触れておいた3群間比較用リアルRNA-seqカウントデータのみを用いて、
おさらい(MacとWinそれぞれの作業ディレクトリの変更など)を含めながらクラスタリングと発現変動解析について述べた。
サンプル間クラスタリングでは、TCCパッケージ中のclusterSample関数のオプションの意味や、Spearman相関係数(r)から1 - rとして距離を定義することで、
数値が小さいほど類似度が高いことを表現できる数式の意味を述べた。最も類似度が高い2つのサンプルに着目し、サンプル間クラスタリング結果の縦軸の高さと、
独立にrから計算した1-r結果が同じであることを示した。クラスタリング結果の解釈として、群間類似度と発現変動解析結果の相関についても述べた。
群間類似度が低いほど2群間比較で得られる発現変動遺伝子(DEG)数が多いことが予想され、実際にそうである(Tang et al., BMC Bioinformatics, 2015)ことを述べた。
上記の検証を兼ねた様々な2群間比較。最も類似度が低いヒト vs. アカゲザルを最初の例として、サブセット抽出のおさらい、
DEG検出結果の解釈上重要なfalse discovery rate (FDR)の説明、緩めのFDR閾値を用いたDEG数の見積もりについて述べた。
M-A plotの説明とDEG検出結果上位のM-A plot上での位置、FDR閾値を変化させたときのM-A plot上でのDEGの分布を説明し、統計的手法の説明のイントロを行った。
FDR 5%条件下で、ヒト vs. アカゲザルの結果は2,488 DEGs、類似度が近いヒト vs. チンパンジーの結果は578 DEGsであり、
クラスタリング結果(全体的な類似度)と同じであることを示した。non-DEG分布の意味を説明すべく、DEGがないことが予想される同一群(ヒト)
の反復データの統計解析結果がDEGがわずかしか検出されないこと、2倍以上の発現変動など倍率変化でDEG検出を行う危険性、3群間比較への展開を述べた。
2015年8月6日(2015.08.06):NGS解析。ChIP-seq(理化学研究所 森岡勝樹先生)
講義資料 (約2MB)
- bowtie2のマッピングで-N0は(マッピングのためのシード領域で許容するミスマッチ数で0または1が選択可能)
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGS速習コース2014
2014年9月1-12日にJST-NBDCと東大農アグリバイオ主催で「バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ)速習コース」が開催されました。 2014年12月に速習コースの動画が統合TVおよびYoutubeから公開されました。 ハッシュタグは#AJACS。
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ)関連:
- NBDCの速習コースサイト(速習コース主催機関)
- カリキュラムを策定したNBDC運営委員会人材育成分科会
- 「NBDCで実施した調査」のバイオインフォマティクス人材育成のためのカリキュラム
- 「バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ)」のPDF (generation-sequencer.pdf)
- 「カリキュラムで習得できる技能」のPDF (learning-skills.pdf)
- 「カリキュラム フロー図」のPDF (flow-diagram.pdf)
- 速習コースアンケート用紙
- PDF版(questionnaire_2014.pdf)
- Microsoft Word版(questionnaire_2014.docx)
- Microsoft Word 97-2003版(questionnaire_2014.doc)
記名回答者のみご利用ください。無記名の方は紙ベースのもので提出お願いします。
メールの添付で 宛てに、件名は「NGS速習コースアンケート」でお願いします。
宛てに、件名は「NGS速習コースアンケート」でお願いします。
計算機環境構築(Linux系):
Linux環境構築手順は大まかに3つの手順からなります。
最低限、VirtualBoxのインストールができていればOKです。VirtualBoxのバージョンは2014年9月の実習では4.3.12以前のものを想定しています。
イメージファイルは、初日に速習コース会場にて、USBメモリで持ち込みPCにコピーすることも可能です。
また、何らかの理由により持ち込みPCにインストールできなかったとしても主催機関であるアグリバイオインフォマティクス所有のインストール済みのノートPC(60台程度あり)をすぐに貸与可能です。
インストール手順(Windows用):install_NGSsokushu_windows.pdf(2014.08.22版;約6MB;西岡 輔 氏作成)
インストール手順(Macintosh用):install_NGSsokushu_macintosh.pdf(2014.08.26版;約3MB;孫 建強 氏作成)
トラブルシューティング(ファイルシステム関連):troubleshooting_NTFS.pdf(2014.08.14版;約1MB;西岡 輔 氏作成)
トラブルシューティング(BIOS関連):troubleshooting_BIOS.pdf(2014.08.15版;約2MB;西岡 輔 氏作成)
- VirtualBoxインストール
- イメージファイルBioLinux.ova (アメリエフ様作成)のダウンロード 7.1GB程度あります。東大有線LANで30分程度かかります。ダウンロードリンクは複数提供しているが、どれか一つだけを利用するとよいです。
- BioLinux.ova (Google Drive1)
- BioLinux.ova (Google Drive2)
- BioLinux.ova (adrive)
- BioLinux.ova (東大アグリバイオ; 非力なので非推奨)
- VirtualBoxへのイメージファイルの導入
補足情報
2014年9月の速習コースで利用したイメージファイルは、Bio-Linux 7をベースに下記のいくつかのプログラムや解析用デモデータを追加したファイルになります(アメリエフ様作成)。 Bio-Linux 8が2014年7月末にリリースされましたが今回の速習ではBio-Linux 7となっております。 イメージファイル中にはBio-Linux 7を含めFIREやngs.plotはすでにインストールされていますので、ユーザが新たにこれらをインストールする必要はありません。 7.1GBというトンデモナイファイルサイズになってしまうのは、デモデータなどを含め一度に1つのファイルとしてダウンロードするためです。 仮想環境の概念については、日本乳酸菌学会誌のNGS関連連載の第1回分PDFが参考になるかもしれません。- Bio-Linux:Field et al., Nat Biotechnol., 2006
- Bio-Linuxに追加でインストール済みのプログラムリスト
計算機環境構築(アプリケーションソフトウェア系):
- Rおよび関連パッケージ MacユーザのヒトもWindows用のものを参照し、ちゃんとパッケージがインストールされているかどうかを確認しておいてください。
- Windows用:R_install.pdf(2014.07.31版;約3MB)
- Macintosh用:R_install_mac.pdf(2014.05.14版;約3MB;孫 建強 氏作成)
- IGV:Thorvaldsdóttir et al., Brief. Bioinform., 2013
速習コース講義資料:
統合TVで公開予定の講義と講義資料PDFです。公開可能なものについて随時更新しています。 以下のような形式で示します。2014年12月4日に統合TVへのリンクを張りました。 速習コース全体を俯瞰できるYoutubeのリストはこちら。
- 日時、「項目番号および項目」、レベル(初級または中級)、形式(講義または実習)
- 講師と所属、統合TVリンク先、講義資料(PDFファイル)
- 講義内容
- 2014年9月1日10:40-12:00、「1-1. OS, ハード構成」、初級、講義
- 中村保一 (DDBJ)、 統合TV(1-1、1-2共通)、講義資料
- コンピューターの基本の理解。必要なスキル。ハードウェア(計算機の歴史、現代のハードウェア構成)。遺伝研スパコンを例に。 ソフトウェア(オペレーティング・システム(OS)、言語・プログラム・ライブラリ)。UNIX OSや、UNIXの構成、OSのご利益。 シェル環境(bash, tcsh, zsh)。スクリプト言語。米国ではPythonが人気。
- 2014年9月1日13:15-14:45、「1-2. ネットワーク基礎」、初級、講義
- 中村保一 (DDBJ)、 統合TV(1-1、1-2共通)、講義資料(追加分)
- インターネット、セキュリティの基本の理解。ネットワーク(よく使うインターネットプロトコル)。インターネットの始まり、TCP/IP。 よく使う通信プロトコル(HTTP, FTP, SSH, SFTP, SMTP/POP)。スパコンへのログインはWindowsはTera Term、Macはターミナルを起動。 外部サーバとのファイル送受信(sftp, Cyberduck, Asperaなど)。DDBJ DRAの話。 データ量の推移や検索の仕方、解析パイプラインも用意しており、主要な生物種ではLinuxを覚えなくてもどうにかなる。 遺伝研スパコンは無料。GNU/Linux。DDBJパイプラインを用いて公開データ納豆菌をVelvetでアセンブルする話。SRAメタデータの説明。
- Tera Term (Windows用)
- Cyberduck(Macintosh用とWindows用)
- DRA
- DDBJ Read Annotation Pipeline:Nagasaki et al., DNA Res., 2013
- DRA search
- 遺伝研スパコンの過去の講習会資料(2012年5月10日のPDFファイル)
- 2014年9月1日15:00-18:15、「1-3. UNIX I」、初級、実習
- 仲里猛留 (DBCLS)、 統合TV、講義資料 (2014.08.29版)
- UNIXの基礎の理解、VirtualBoxとBioLinux環境構築。UNIXはOSであり、Windows 7やMacOS Xと同じようなもの。 マルチユーザー、マルチタスク、super user, root。Linuxは個人で使うレベルのUNIX。 ディストリビューション(Debian系、RedHat系、OpenSuSe)。カーネル(kernel)とシェル(shell)。 BioLinuxの起動と終了、画面説明、ターミナルの起動。ディレクトリとファイル。 環境構築がほぼ全員できていたので、STAFFおよびTAで個別対応。順調に進んだため、9/2分の講義を引き続いて行った。 lsコマンド、オプションの使い分け。プロンプトは自在に設定可能。引数はカタマリごとなら入れ替えてもよい。 ディレクトリ構造。cdコマンド。ホームディレクトリは「ちるだ」、カレントディレクトリは「どっと」、 一つ上のディレクトリは「どっとが二つ」、ディレクトリの区切りは「/」。絶対パスと相対パス。 基本コマンド:ls, cd, pwd, mv, cp, rm, mkdir, cat, more/less, head/tail。 質疑応答で様々なオプションやTipsを共有。rmコマンドの-fや-rオプション。headコマンドの-15オプションで15行分見られるとか。 「CTRL + L」ボタンでターミナルをリフレッシュ。lsコマンドでワイルドカードの意味を説明。 「/home/admin1409/amelieff/perl/pl_answer」ディレクトリ上で「ls -l perl?.pl」とか「ls -l perl*.pl」での挙動の違いを確認。 ゲストOS上のコマンドのコピーはホストOSの「CTRL + C」などはだめなので、右クリックでやるべし。 ホストOS (WindowsやMacintosh)上のファイルをゲストOS (BioLinux)上にコピーするのはドラッグアンドドロップで可能。 その逆はデフォルトでは不可。日本語を含む20140911_4-4_kawaoka.txtをBioLinux上でみると文字化けするが、 下記のようにiconvコマンドで「iconv -f SJIS 20140911_4-4_kawaoka.txt > hoge.txt」で解決可能。 nkfコマンドがあればそれでも解決できるらしい。
- 2014年9月2日10:30-18:15、「1-3. UNIX I」、中級、実習
- 仲里猛留 (DBCLS)、 統合TV、講義資料 (2014.09.01版)
- 権限(Permission)、「Read, Write, eXecute」、「ユーザ, グループ, その他」。chmod, su, sudoコマンド。 ~/genome/yeast.ntを用いてgrep, sort, uniqコマンドを実行。 「grep GAATCC genome/yeast.nt」、「grep ^GAA genome/yeast.nt」、「grep GAA$ genome/yeast.nt」など。 パイプ(縦棒)の話。「grep GAATCC genome/yeast.nt | wc -l」などいろいろ。より高機能なegrepもある。 正規表現、シングルクォーテーションとダブルクォーテーションの違い。 「echo '$SHELL' 任意のファイル名」と「echo "$SHELL" 任意のファイル名」。 「wc –l genome/yeast.nt」、「sort genome/yeast.nt | uniq | wc –l」、「sort genome/yeast.nt | uniq –c | sort –n」、 「sort genome/yeast.nt | uniq –c | sort –rn | head -3」、 「sort genome/yeast.nt | uniq –c | sort –rn | head -3 | less –S」。 リダイレクトでファイルに保存。「sort genome/yeast.nt | uniq > genome/yeast.uniq.nt」。 「sort genome/yeast.nt | uniq」、画面表示を途中で止めたい場合は「CTRL + C」。 Cufflinksというプログラムをwgetして*.tar.gzファイルが存在するところからの代表的なインストール例。 「tar xvzf cufflinks-2.2.1.tar.gz」、「cd cufflinks-2.2.21」、「whereis make」。 今回の実習環境ではnkfコマンドが入ってなかったので、みんなで「sudo apt-get install nkf」、 「apt-cache search nkf」。そして文字化けしていたファイルの文字コード変換を実行。 「nkf 20140911_4-4_kawaoka.txt > hoge2.txt」。パスの話。「echo $PATH」。 デフォルトで「bwa」と打つとbwa ver. 0.6.1-r104となるが、ホームディレクトリ以下にbwa ver. 0.7.10が既にインストールされている。 bwa最新版を「bwa」で利用したいときにパス指定をしたり、lnコマンドでシンボリックリンクを作成したりする。 エディタの紹介。viやemacsがあるが、emacsはターミナルで利用可能な「CTRL + e」や「CTRL + a」などが共通。 マルチタスクの実例。「CTRL + z」で終了、「Jobs」で確認、「bg %1」や「fg %2」など。 「ssh admin1409@localhost」で、自分自身のマシンに新たにログイン。「screen」コマンド。
- Linuxコマンド集:20140902_1-3_nakazato_linux.pdf
- エディタ(emacs, vi)のキーバインド集:20140902_1-3_nakazato_editor.pdf
- wgetとunzip実例:20140902_1-3_wget_unzip.txt (2014.09.02版)
- 要望:「/home/admin1409/amelieff/perl/pl_answer/」には多数のperlプログラムがあるが、 例えば*.plの前の数値を自動的に+10したファイル名に変更するやり方を知りたい。
- やり方1(長尾 吉郎 氏 提供情報):
- ファイル名変更perlプログラム:20140902_rename.pl (2014.09.03版)
- 実行例:20140902_rename1_commands.txt (2014.09.03版)
- 解説:test1ディレクトリ中に5つのファイル(perl2.pl, perl10.pl, perl_kk.pl, perl_87.pl, perl_87.txt)が存在する条件下で、20140902_rename.plを実行しています。 「perl 20140902_rename.pl *.pl」は、拡張子が*.plというファイルのみ入力(それゆえ該当するのはperl2.pl, perl10.pl, perl_kk.pl, and perl_87.plの4ファイル)として、 拡張子の直前の数値を+10してrenameする20140902_rename.plという名前のperlプログラムを実行する、という意味です。 実行後にlsすると「20140902_rename.pl perl12.pl perl20.pl perl_87.txt perl_97.pl perl_kk.pl」となっていて、 拡張子の直前が数値となっているファイルのファイル名が変更されていることがわかります。たとえばperl2.plはperl12.plという名前に変更されています。 それゆえ、例えば実行するためのperlプログラム名が20140902_rename.plではなく20140902_rename1.plだと、このファイル名も変更されるという特徴があります。
- やり方2(甘利 典之 氏 提供情報。もらったファイルを門田が変更したことに起因する間違いの可能性あり):
- ファイル名変更シェルスクリプト:20140902_rename.sh (2014.09.03版)
- 実行例:20140902_rename2_commands.txt (2014.09.03版)
- 解説:test2ディレクトリ中に5つのファイル(perl2.pl, perl10.pl, perl_kk.pl, perl_87.pl, perl_87.txt)が存在する条件下で、20140902_rename.shを実行しています。
「sh 20140902_rename.sh」は、拡張子が*.plというファイルのみ入力(それゆえ該当するのはperl2.pl, perl10.pl, perl_kk.pl, and perl_87.plの4ファイル)として、
拡張子の直前の数値を+10してrenameする20140902_rename.shという名前のperlプログラムを実行する、という意味です。
実行後にlsすると「20140902_rename.sh perl12.pl perl20.pl perl_87.txt perl_97.pl perl_.pl」となっていて、
拡張子の直前が数値となっているファイルのファイル名が変更されていることがわかります。たとえばperl2.plはperl12.plという名前に変更されています。
一番最後のperl_plは、元がperl_kk.plだったものです。これは拡張子の直前が数値ではないので想定外のことが起こっているということになります。
もちろん条件分岐をすればどうにかなりますが、そういったものをつけていって想定外にどこまで対応するかどうかは状況次第です。
プログラムはそのままで、拡張子の直前がかならず数値にするようにファイル名作成段階でしておくというのが現実的な対応策だと思います。
- 2014年9月3日10:30-18:15、「1-4. スクリプト言語 (シェルスクリプト)」、中級、実習
- 服部恵美 (アメリエフ)、 統合TV、講義資料 (2014.09.08版)
- シェルスクリプトの基礎(変数、引数、条件付き処理、繰り返し処理、標準出力と標準エラー出力、シバンなど)。 変数を使うと値を一元管理できる。変数にしたほうがいいのは、入力ファイル名(リファレンスゲノム、塩基配列のファイル名)とか。 変数名に使わないほうがいいものは「変数名 予約語」などで検索するとよい。 引数を使うとスクリプトの中身をいちいち書き換えなくてよくなる。$1や$2以外に「$0」もある。 例えば、$0はUsageのところなどで利用される。条件付き処理でmkdir。複数の条件を指定したいときは「-aや-r」を利用。 bashのif文で使用する-eの意味は、指定した名称のファイルが存在するかではなく、 指定した名称のディレクトリまたはファイルが存在するかであろう(中杤昌弘 氏 提供情報)。 「bash -n ファイル名」でシェルのコーディング確認ができる。echoで実行コマンドを出力させて結果をわかりやすくする。 標準出力と標準エラー出力。「>&2」が標準エラー出力。「bash test7.sh 1>log 2>err」は、 標準出力はlogというファイルに出力し、標準エラー出力はerrというファイルに出力する。 プログラム内部を理解しやすくするため、スクリプト中に#から始まるコメントを入れた方がわかりやすい。 「シバン」というのは、一般的にプログラムの一行目に書く。 このプログラムはシェルスクリプトである、ということを示す場合は「#!/bin/bash」と書く。 こうすることで例えば「bash test.sh」と書く代わりに「./test.sh」で済む。 sequence logos (weblogo)の紹介をして、mature.faやhairpin.faを入力としてレベルに応じた最終課題(mkdir, grep, weblogoなど)。
- テストデータ:hairpin.fa.zip (miRNA前駆体配列)
- テストデータ:mature.fa.zip (成熟miRNA配列)
- 要望:実習6の発展課題周辺のコードで、任意の数のディレクトリを引数で与えて作成したい。
- やり方1(田中洋子 氏 提供情報):
- シェルスクリプト:20140903_mkdir1.sh (2014.09.04版)
- 実行例:20140903_mkdir1_commands.txt (2014.09.04版)
- 解説:「bash 20140903_mkdir1.sh uge hogege hangya」と打つと、uge, hogege, hangyaという三つのディレクトリが作成されます。
- やり方2(長尾 吉郎 氏 提供情報):
- シェルスクリプト:20140903_mkdir2.sh (2014.09.04版)
- 実行例:20140903_mkdir2_commands.txt (2014.09.04版)
- 解説:引数で指定した文字列を与えると左側に"CITY_"という文字列が付加された三つのディレクトリが作成されます。
例えば、「bash 20140903_mkdir2.sh uge hogege hangya」と打つと、CITY_uge, CITY_hogege, CITY_hangyaという三つのディレクトリが作成されます。
- その他1(孫建強氏 提供情報):上記20140903_mkdir*.shを作成する際に、しばらく「bash -n 20140903_mkdir*.sh」でエラーが出るという問題に苦悩しました。 原因はWindows OS上で作成したものをBioLinux上でダウンロードして使おうとしていたために、"\r"が原因と推認されるエラーメッセージが出ていました。 これは結論としてはOS間の改行コードの違いによる問題で、同じプログラムをBioLinux上で作成し直すとうまく動きました。 こういうことはしょっちゅうありますのでご注意ください。
- その他2(田中洋子 氏 提供情報):改行コードが 「CR+LF」になっているシェルをviやemacsで開くと「^M」と表示されていて識別可能。 WindowsとLinux間でファイルサイズが違っているのは改行コードの違いによる。 具体的には、「CR」の1バイトの違いが行数分だけファイルサイズの違いに表れているのがわかる。 WinSCPのようなファイル転送ソフトだとデフォルトのテキストモードでWindowsとLinuxOS間でテキストファイルを転送すると 「賢く」改行コードを変換してくれるため、特に気にする必要はないが、Windows上で作成されたテキストファイルを BioLinux上で直接wgetコマンドで取得するとプログラムが動かないということになるので注意。 ただし、賢く変換してくれるがゆえに、大容量のFASTQファイルなどテキストモードで転送しようものなら 延々頑張って改行コードを変換してくれて転送が全然終わらないということになるのでそのあたりは注意が必要。
- その他3(中杤昌弘 氏 提供情報):
- シェルスクリプト:20140903_mkdir3.bsh (2014.09.07版)
- 実行例:20140903_mkdir3_commands.txt (2014.09.07版)
- 解説:最初の2個の引数で指定した名前のディレクトリを作成するスクリプト。3個目以降引数は別の用途に使用できる。 例えば、「bash 20140903_mkdir3.bsh uge hogege hangya」と打つと、最初の2つの引数で指定したディレクトリugeとhogegeが作成され、 3つめの引数で指定したhangyaは作成されないことがわかります。拡張子はshでなくてbshでもよいのです。
- その他4(中杤昌弘 氏 提供情報):
- シェルスクリプト:20140903_mkdir4.bsh (2014.09.07版)
- 実行例:20140903_mkdir4_commands.txt (2014.09.08版)
- 解説:bashのif文で使用する-eの意味は、指定した名称のファイルが存在するかではなく、 指定した名称のディレクトリまたはファイルが存在するかであることを検証するためのスクリプト。 ホームディレクトリ直下に存在するgenomeを引数として与えている実行例を行うと、確かに-eオプションがTRUEとなっていることがわかる。
- 2014年9月4日10:30-18:15、「1-4. スクリプト言語 (Perl)」、中級、実習
- 服部恵美 (アメリエフ)、 統合TV、講義資料 (2014.09.08版)
- Perlの基礎(変数、配列、引数、ハッシュ、条件付き処理、繰り返し処理、ファイル入出力、シバン、正規表現など)。 一般に、Perlのほうがシェルスクリプトよりも複雑な処理に向いていると言われる。 例えば、ファイル形式の変換や結果ファイルの独自解析など。達成目標は、hairpin.faの特定のRNAの配列を 切り出して別のファイルに書き出すPerlプログラムを書けるようになる。Perlの記載方法。値を出力するにはprintを実行、 文字列はダブルクォートかシングルクォートで囲む、行の末尾に「;」をつける、全角記号や全角空白文字は使えない。 シェルスクリプト同様、Perlでも「変数」を使うことができる。「$変数」と書くと、変数に入っている値を呼び出すことができる。 最初に変数が出てくるときには「my」をつける。シェルスクリプトと違って、代入の「=」の前後に空白が入っても大丈夫。 Perlのprintはシェルスクリプトのechoと違って最後が改行されない。 そのため、改行したい場合は明示的に「\n」と改行コードを書く必要がある。 「配列」を使うと、たくさんの値をまとめて扱うことができる。配列からの値の取り出し方。 「添字」は0から始まる。push関数で配列に値を追加。配列の要素数の調べ方(Rのlength関数に相当)。 配列を結合して文字列にするのはjoin関数。文字列を分割して配列にするのはsplit関数。 Perlでも引数が使える。Perlでは、引数を「@ARGV」という名前の専用の配列で受け取る。 ハッシュの話。「$ハッシュ名{キー}」でハッシュから値を取り出す。配列とハッシュの違い。 Perlでも条件付き処理や繰り返し処理が可能。シェルスクリプトと書き方が似ているが微妙に異なるので注意。 条件付き処理は、if-elsif-else構文を使用。Perlの比較演算子。複数の条件指定。 Perlの繰り返し処理にはforeach, for, whileなどがある。繰り返し処理でハッシュにアクセス。 繰り返し処理の中で次の要素スキップするにはnextを使用。繰り返し処理自体を中止するにはlastを使用。 {}囲んだ範囲をスコープと呼ぶ。ファイル入出力「use strict;」と「use warnings;」でバグを見つけやすくする。 #で始まる行はコメント扱いとなり、処理に影響しない。 Perlプログラムの1行目に「#!/usr/bin/perl」を記述すると、このファイルがPerlスクリプトであることが明示的になる。 正規表現は文字列のパターンを表現する方法であり、Perlでは、正規表現を//で定義する。 「~s/正規表現パターン/置換文字列/オプション」で、正規表現を用いた文字列の置換を行うことができる。 最終課題は、hairpin.faの塩基配列行を1行にするなど。
- miRBase
- テストデータ:hairpin.fa.zip (miRNA前駆体配列)
- テストデータ:mature.fa.zip (成熟miRNA配列)
- 解答例の修正版:perl10.pl (2014.09.24版)
- 解説:17行目のところを「$id2seq{$id} = $_;」から「$id2seq{$id} .= $_;」に変更しています。
- Tips(服部恵美 先生 提供情報;2014.09.24, 18:26):
- OS間の改行コードの違いについて、例えばWindowsで作成したファイルの改行コードは、 Perlプログラム中で以下のように、chompする前に\rを除去する処理を入れればよい。
s/\r//g;
chomp;
- AAIndex:Kawashima et al., Nucleic Acids Res., 2008
- BLAST:Altschul et al., J Mol Biol., 1990
- FASTA:Pearson and Lipman, Proc Natl Acad Sci U S A., 1988
- GGRNA:Naito and Bono, Nucleic Acids Res., 2012
- FASTQ形式
- SRA/SRA-lite形式
- SAM/BAM形式:Naito and Bono, Nucleic Acids Res., 2012
- VCF (Variant Call Format)形式
- Wiggle (Wig)形式
- GTF/GFF形式
- HsDJ1.pep.fa
- BmDJ1.pep.fa
- 講義資料中のコマンド群
- EPAS1-001.nt.fa
- EPAS1-007.nt.fa
- 講義資料中のコマンド群
- 講義資料中のコマンド群:DBの準備とBLASTの実行
- 講義資料中のコマンド群:オプションを変えてみる
- 講義資料中のコマンド群:例2-2
- 例2-3
- Local BLASTの使い方の統合TV
- AJACS名古屋
- GGRNA:Naito and Bono, Nucleic Acids Res., 2012
- ggrnaapi.pl関連のコマンド群
- count.pl関連のコマンド群
git clone https://github.com/bonohu/AJACS47 (実習用PCにはgitは入ってなかった...)
wget https://github.com/bonohu/AJACS47.master.zip (gitが入っていない場合はこちら)
dottup -asequence HsDJ1.pep.fa -bsequence BmDJ1.pep.fa -wordsize 4
dottup -asequence HsDJ1.pep.fa -bsequence BmDJ1.pep.fa -wordsize 4 -graph png (これでpng形式ファイルに保存できる。これだとファイル名は指定できない。by ツイート情報)
dottup -asequence HsDJ1.pep.fa -bsequence BmDJ1.pep.fa -wordsize 4 -graph pdf (これでpdf形式ファイルに保存できる。これだとファイル名は指定できない。by ツイート情報)
dottup -asequence HsDJ1.pep.fa -bsequence BmDJ1.pep.fa -wordsize 4 -graph png -goutfile hoge (これでhoge.pngというpng形式ファイルで保存できる。by ツイート情報)
dottup -asequence HsDJ1.pep.fa -bsequence BmDJ1.pep.fa -wordsize 10
dottup -asequence HsDJ1.pep.fa -bsequence BmDJ1.pep.fa -wordsize 2
needle HsDJ1.pep.fa BmDJ1.pep.fa
water HsDJ1.pep.fa BmDJ1.pep.fa
less np_009193.needle
less np_009193.water
dottup -asequence EPAS1-001.nt.fa -bsequence EPAS1-007.nt.fa
needle EPAS1-001.nt.fa EPAS1-007.nt.fa
water EPAS1-001.nt.fa EPAS1-007.nt.fa
less enst00000263734.needle
less enst00000263734.water
cd /home/admin1409/genome
makeblastdb -in yeast.aa -dbtype prot -hash_index
makeblastdb -in yeast.nt -dbtype nucl -hash_index
blastp -query ~/HsDJ1.pep.fa -db yeast.aa -num_threads 4 > HsDJ1vsyeast.aa.txt
blastp -query ~/HsDJ1.pep.fa -db yeast.aa -num_threads 4 -evalue 1
blastp -query ~/HsDJ1.pep.fa -db yeast.aa -num_threads 4 -outfmt 6
blastp -help | less
tblastx -query ../EPAS1-001.nt.fa -db yeast.nt -num_threads 4 -evalue 1 | less
perl ggrnaapi.pl | less
perl ggrnaapi.pl | sort | uniq | less
perl ggrnaapi.pl | perl count.pl
perl ggrnaapi.pl | perl count.pl | sort -rn -k2
- 統合TV:Kawano et al., Brief Bioinform., 2012
- バイオ系DB
- NCBI
- Ensembl:Zerbino et al., Nucleic Acids Res., 2018
- Biomart:Kasprzyk A., Database, 2011
- The UCSC Genome Browser:Kuhn et al., Brief. Bioinform., 2013
- IGV:Thorvaldsdóttir et al., Brief. Bioinform., 2013
- NBDCとDBCLSと統合DB
- Integbio データベースカタログ
- 生命科学データベース横断検索
- 生命科学系データベース アーカイブ
- 統合データベース講習会 AJACS, MotDB
- 新着論文レビュー
- 領域融合レビュー
- Allie: 生命科学分野の略語/展開形検索
- inMeXes: 逐次PubMed/MEDLINE表現検索
- RefEx
- GGRNA:Naito and Bono, Nucleic Acids Res., 2012
- GGGenome
- テキスト比較ツール difff《デュフフ》
- DBCLS SRA:Nakazato et al., PLoS One, 2013
- ライフサイエンスQA
- togo picture gallery
- GGRNAとGGGenomeを扱えるステキなRパッケージgenesearchr (by ツイート情報)
- 9/8-9の2日間で用いる全データファイル:hoge.zip (2014.08.28版)
- Rコード:rcode_20140908.txt (20140826,14:18版)
- Macのディレクトリの変更は、メニューバー「その他」から「作業ディレクトリの変更」で出来る。(by ツイート情報)
- Windows OSでコードの全選択をする場合は、コードの枠内で「CTRL + ALT + 左クリック」以外に「トリプルクリック」でもよい。 Macintosh OSは講習会中も全選択のやり方は分からずじまい。 Macユーザのあるヒトは「ドラッグするのが面倒なので、開始位置で一度クリックして終了位置までスクロールしてからShift押しながらクリックして広範囲選択してます (by ツイート情報)」。
- スライド24の条件判定時に括弧をつけているが、講師的にはただの見栄え的なものと説明した。 しかしPerlの演算子と優先順位のようなものがRにもある(by ツイート情報)。 演算子の優先順位に関する記述はR Tipsの28. 演算子にもある(by ツイート情報)。
- is.element関数での条件判定時に、Windows OSとMacintogh OSの多数派ではas.characterの有無で結果は変わらない(R ver. 3.1.0)が、
Macユーザの一人はas.characterをつけないとエラーが出て、もう一人はas.characterをつけるとエラーが出た。
sessionInfo()は見ていないのでバージョンがちょっと違うのかもしれない。
- source関数実行時に利用:rcode_translate.txt
- Macのヒトで作業スペースのロード時に、.RDataファイルが隠しファイルとなっていて場所が分かりづらかった(by ツイート情報)という点は重要な指摘。
- Rコード:rcode_20140909.txt (2014.08.28版)
- Rコード:rcode_translate2.txt
- Rコード:rcode_translate3.txt
- DNAStringSetに格納されてる各配列は、fasta[[1]]等とするか、namesで指定して、 fasta$gene1等とすればDNAStringとして得られるので、その後の解析によっては、文字列で得るよりも扱いやすい場合もあるかも(by ツイート情報)。
- オブジェクトの消去はrm()後に、gc()というコマンドを2回打ち込んでメモリーを解放するのがお作法。
- パッケージ中のオブジェクト評価関連(小杉孝嗣 氏 提供情報):
- Bsgenome.Hsapiens.UCSC.hg19のようなゲノム配列パッケージから、パッケージ名と同じ名前のオブジェクトを抽出する目的で
「eval(parse(text=tmp))」のように文字列tmpをオブジェクトとして取り扱っているが、基本的にはtmpの中身は一つのオブジェクトしかないという前提で門田らは作成していた。
しかし、実際には複数個の文字列が含まれる場合がある。例えばイントロ | 一般 | 配列取得 | ゲノム配列 | BSgenome
の5.の途中で用いているtmpオブジェクト中には、当初想定していたパッケージ名と同じ"BSgenome.Hsapiens.UCSC.hg19"以外に、"Hsapiens"という文字列も存在する。
実際に取り出されているのは、tmpオブジェクト中の最後の要素(この場合、"Hsapiens")である。
ここではたまたまこの2つのオブジェクトの中身が同じだったので問題なく"Hsapiens"の中身が当初想定していた"BSgenome.Hsapiens.UCSC.hg19"と同じだったのでうまくいっているが、
そうではないことも十分考えられるので気にしておかねばならない。
- writeFastq関数は同名ファイルが存在する場合は書き込み権限がないとエラーが出るが、writeXStringSet関数は同名ファイルの有無に関わらず 上書きするらしい(by ツイート情報)。
- オブジェクトサイズについて(土居主尚 氏 提供情報):
- object.sizeという関数で、どのオブジェクトがどの程度メモリを消費しているかを調べることができる。
- fastq形式ファイル:1K_ERR038793_1.fastq
- bam/sam形式ファイル:1K_ERR038793.bam
- vcf形式ファイル:1K_ERR038793_sort.vcf
- fasta形式ファイル:genome.fa
- SAMtools
- IGV
- IGV関連(細谷将 氏 提供情報):
- リファレンスゲノムを指定して立ち上げるのは「igv.sh -g genomeID」でできる。例えばhg19なら「igv.sh -g hg19」
- IGV関連(田中洋子 氏 提供情報):
- IGVトップ画面の見出し欄は非常にきれいに大中小項目分類されていてシンプルでよいが、 最初は自分の知りたい情報を見つけにくいヒトもいるかもしれない。その場合、慣れるまでは「Documents」-「IGV User Guide」 以下をすべて開いて使うとよい。
- IGVのコマンドスタート説明
- ゲノムは講師の先生の環境にはmm9 があったが、講習環境では{home}/igv/genomes にはhg18しかなかった。 しかし上記説明箇所のリンク[the genome registry]を開いてmm9.genome を見つけてwgetでダウンローできる。 この後の講習で出てくるかもしれないが、.genome前がIDです。例えば、mm9.genomeの場合はIDがmm9となる。 説明にもあるように、今はゲノムIDだけでなく参照に使いたいゲノムのファイル指定もできる。
- 講習環境のIGV画面からのリファレンスゲノムの切り替えをやってみましたが、何度やってもいつまでたっても終わらない場合は、 wgetで.genomeを取得してから切り替るほうがさくっとできてよい。おそらくIGVサイトからのダウンロードと.genome作成をやっているのであろう。
- IGVの状態は、ホーム直下のigvディレクトリの/home/admin1409/igv/prefs.properties に随時保存されるので、終了時の内容で次回起動する。 例えば、起動時に表示されるリファレンスゲノムはDEFAULT_GENOME_KEYで決まる。この設定ファイルの存在を知っているとIGVの挙動理解につながる。
- IGVのバッチファイルはサイトの例同様、見たい領域が何か所もある時に利用していて便利。
各領域について位置指定+スナップショットで保存のバッチ作成。バッチの実行はIGVメニューからもコマンドからも実行できる。
ただしスナップショットなので、たしか下スクロールになってしまう箇所までは保存できなかったように思います。
- BioLinux上で「R」と打ち込むとRが起動します。「>」というプロンプトが出ている状態で以下をコピペ。(2014.09.18更新)
- 3つのパッケージがインストールできているかどうかの確認。 BioLinux上で「R」と打ち込むとRが起動します。「>」というプロンプトが出ている状態で以下をコピペ。 エラーメッセージが出ている人は挙手!。特に問題ないヒトはRの終了。終了は「q()」。 このとき、「Save workspace image? [y/n/c]:」と聞かれるが「n」でよい。
- 全データファイル(約410MB):kawaoka.zip (2014.09.18版)
- コピペ用コマンド集:20140911_4-4_kawaoka.txt (2014.09.18版)
- kawaoka_test.sh (20140831,13:44版)
- chr17_base.1.bt2 (約30MB)
- chr17_base.2.bt2 (約20MB)
- chr17_base.3.bt2 (約1KB)
- chr17_base.4.bt2 (約20MB)
- chr17_base.rev.1.bt2 (約30MB)
- chr17_base.rev.2.bt2 (約20MB)
- chr17_cMyc.sam (約260MB)
- chr17_cMyc_uniq_sorted.bam (約40MB)
- chr17_Input.sam (約240MB)
- chr17_Input_uniq_sorted.bam (約35MB)
- chr17_Oct4.sam (約240MB)
- chr17_Oct4_uniq_sorted.bam (約35MB)
- chr17.fa (約80MB)
- cMyc_top100.txt (約80KB)
- Oct4_top100.txt (約60KB)
- SRR445816_fastqc.zip (約120KB)
- uni.fastq (約5MB)
- Gene Expression Omnibus (GEO)
- BioLinux上でエクセルシート的な解析を行うのは「LibreOffice Calc」というものであるが、使い慣れていないヒトにとっては苦行。
使い慣れたホストOS上のEXCELなどでやりたい場合には、VirtualBoxの「設定」ー「共有フォルダ」を設定して利用するとよい(by ツイート情報)。
source("http://www.bioconductor.org/biocLite.R")#おまじない
biocLite("ShortRead") #Bioconductor中にあるShortReadパッケージをインストール
biocLite("BSgenome") #Bioconductor中にあるBSgenomeパッケージをインストール
biocLite("doMC") #Bioconductor中にあるdoMCパッケージをインストール
コピペ中にいろいろメッセージが出る。
「>」というプロンプトが出ると一通りの処理が終わり、次のコマンド入力待ち状態になる。
その状態で「library(ShortRead)」と打ち、何らかのエラーメッセージが出ていないことを確認。
同様に「library(BSgenome)」と打ち、何らかのエラーメッセージが出ていないことを確認。
終了は「q()」。このとき、「Save workspace image? [y/n/c]:」と聞かれるがとりあえず「n」でよい。
library(ShortRead)
library(BSgenome)
biocLite("doMC")
library(ShortRead)
library(BSgenome)
biocLite("doMC")
個別のファイルはこちら:
- コマンド例:Reseq_command.txt
書籍 | トランスクリプトーム解析 | について
2014年4月に(Rで)塩基配列解析および(Rで)マイクロアレイデータ解析 を体系的にまとめた以下の書籍が出版されました。ここでは(Rで)塩基配列解析に関連したRNA-seq部分の書籍中のRコードを章・節・項ごとに示します。
なるべく書籍中の記述形式に準拠しますが、例えばp72中の最初のsetwd("C:/Users/kadota/Desktop")というコマンドを忠実に実行してもエラーが出るだけです。 これはkadotaというヒトのPC上でのみ成立するコマンドだからです。 ここで利用しているsetwd関数は作業ディレクトリの変更に相当し、「ファイル」−「ディレクトリの変更」でデスクトップに移動することと同義です。 したがって、このページ全体で統一的に使っているように、ディレクトリの変更作業自体はR Gui画面左上の「ファイル」メニューを利用することとし、 setwd関数部分の記述は省略します。いくつかの数値の違いは、Rのバージョンの違いもあるでしょうし、取得先のデータベース側のバージョンの違いにもよると思います。 したがって、「得られるファイル」や「入力ファイルのリンク先のもの」と「手持ちの入力ファイル」が多少異なっていても気にする必要はありません。
- 門田幸二著(金明哲 編), シリーズ Useful R 第7巻 トランスクリプトーム解析, 共立出版, 2014年4月. ISBN: 978-4-320-12370-0
書籍 | トランスクリプトーム解析 | 1.1 はじめに
シリーズ Useful R 第7巻 トランスクリプトーム解析のp1-3です。
p1:
- ヒトゲノム:UCSCの大元
- ヒトゲノム:Feb. 2009(hg19, GRCh37)(書籍執筆時のバージョンはこれ)
- ヒトゲノム:Dec. 2013(hg38, GRCh38)(2014年5月9日現在の最新バージョン)
- ジストロフィン(dystrophin):Monaco et al., Nature, 1986
- ジストロフィン(dystrophin):NC_000023.10(書籍執筆時のバージョンはこれ)
- ジストロフィン(dystrophin):NC_000023.11(2014年5月9日現在の最新バージョン)
p2:
- FANTOM:Functional Annotation of the mammalian genomeの略
- FANTOM5:FANTOM Consortium et al., Nature, 2014
- ENCODE:Encyclopedia of DNA Elementsの略
- ENCODE:ENCODE Project Consortium et al., Nature, 2012
- GENCODE:Harrow et al., Genome Biol., 2006
p3:
書籍中での言及はないが、DBCLS SRAも便利なので追加しています。
- GEO:Barrett et al., Nucleic Acids Res., 2013
- SRA:Sequence Read Archiveの略
- DDBJ SRA(DRA):Kodama et al., Nucleic Acids Res., 2012
- DBCLS SRA:Nakazato et al., PLoS One, 2013
書籍 | トランスクリプトーム解析 | 1.2.1 原理(Affymetrix 3'発現アレイ)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp3-5です。
p3:
- GEO:Barrett et al., Nucleic Acids Res., 2013
- ヒト用:GPL570(Affymetrix Human Genome U133 Plus 2.0 Array)
- マウス用:GPL1261(Affymetrix Mouse Genome 430 2.0 Array)
- ラット用:GPL1355(Affymetrix Rat Genome 230 2.0 Array)
p5:
- アノテーションファイル(GPL570-13270.txt)は、GPL570の「Download full table...」ボタンを押すことでダウンロード可能。
- GGRNA:Naito and Bono, Nucleic Acids Res., 2012
- Probe Search
- GGRNAを用いたHumo sapiens(human)中の1552263_atの検索結果
書籍 | トランスクリプトーム解析 | 1.2.2 最近の知見
シリーズ Useful R 第7巻 トランスクリプトーム解析のp5-8です。
p6:
- GenBank:Benson et al., Nucleic Acids Res., 2014(書籍執筆時は2013だったが、2014の原著論文が出たのでそちらに変更しています)
- RefSeq:Pruitt et al., Nucleic Acids Res., 2014(書籍執筆時は2012だったが、2014の原著論文が出たのでそちらに変更しています)
- エクソンアレイ:GPL5188(Affymetrix Human Exon 1.0 ST Array)
- トランスクリプトームアレイ:GPL17585(Affymetrix Human Transcriptome Array 2.0)
- GEO:Barrett et al., Nucleic Acids Res., 2013
- ArrayExpress:Rustici et al., Nucleic Acids Res., 2013
- Expression Atlas:Petryszak et al., Nucleic Acids Res., 2014(書籍には掲載していなかったが有用だと思われるので追加)
書籍 | トランスクリプトーム解析 | 2.2.1 生データ(プローブレベルデータ)取得
シリーズ Useful R 第7巻 トランスクリプトーム解析のp36-38のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p36の網掛け部分:
作業ディレクトリがデスクトップという前提です。有線LANで数分程度かかりますのでご注意ください。
param <- "GSE30533" #入手したいIDを指定 library(ArrayExpress) hoge <- getAE(param, type="raw", extract=F)
p36下:
作業ディレクトリ(デスクトップ)上に、多数の他のファイルが存在する場合には書籍中のlist.files()実行結果と見栄えが異なります。
getwd() list.files()
p37上:
圧縮ファイル解凍後に行います。
list.files()
p37の網掛け部分:
kadotaに相当する部分は人それぞれです。Macintoshのヒトは、それ以外の場所でも違いがあるかもしれません。
list.files(path=getwd()) list.files(path="C:/Users/kadota/Desktop") list.files(path = "C:/Users/kadota/Desktop")
p37下:
p36で指定した入手したいGEI IDが異なれば、当然E-GEOD-30533.raw.1に相当する部分も変わります。
getwd() list.files(path = "C:/Users/kadota/Desktop/E-GEOD-30533.raw.1") getwd() list.files(recursive=TRUE)
p38:
?list.files ?getAE
書籍 | トランスクリプトーム解析 | 2.2.2 データの正規化(基礎)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp39-45のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"に移動し以下をコピペ。
p39:
作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提です。
getwd()
p40の網掛け部分(上):
hoge1.txtと同じものができていると思います。
out_f <- "hoge1.txt" #出力ファイル名を指定してout_fに格納 library(affy) #パッケージの読み込み hoge <- ReadAffy() #*.CELファイルの読み込み eset <- mas5(hoge) #MAS5を実行し、結果をesetに保存 write.exprs(eset, file=out_f) #結果をout_fで指定したファイル名で保存
p40下:
list.files() dim(exprs(eset))
p40の網掛け部分(下):
exprs(eset)[,2] #2列目のデータを表示 exprs(eset)[,4:7] #4列目から7列目のデータを表示 exprs(eset)[,c(1,6,9)] #1,6,9列目のデータを表示 exprs(eset)[1:3,1:2] #1,2列目のデータの最初の3行を表示 head(exprs(eset)[,1:2], n=3) #1,2列目のデータの最初の3行を表示(上と同じ結果) head(exprs(eset)[,1:2]) #1,2列目のデータの最初の6行を表示(デフォルトは6)
p41:
colnames(exprs(eset))
colnames(exprs(eset)[,4:7])
colnames(exprs(eset)[,c(1,6,9)])
ccc <- c("GSM757155_-Fe_short_27.CEL", "GSM757160_control_28.CEL")
exprs(eset)[2:4,ccc] #cccで指定した列の2-4行目のデータを表示
rpr <- "1367453_at"
exprs(eset)[rpr,ccc] #ccc列のrprで指定したprobeset IDデータを表示
param <- "1369078_at"
exprs(eset)[param, 1] #1列目のparamで指定したprobeset IDデータを表示
new_colname <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep=""))
new_colname
colnames(exprs(eset)) <- new_colname
exprs(eset)[2:4,ccc] #cccで指定した列の2-4行目のデータを表示
ccc
exprs(eset)[2:4,] #2-4行目のデータを表示
p42:
作業ディレクトリがデスクトップという前提です。CELファイルがないことがわかっているディレクトリ上で読み込みを行った場合の結果を示しています。 ディレクトリの変更を忘れると、うまくいってしまうのでご注意ください(笑)。
library(affy) #パッケージの読み込み hoge <- ReadAffy() #*.CELファイルの読み込み data <- exprs(eset) #exprs(eset)をdataに代入 boxplot(data) # 1)まずはそのままboxplot boxplot(data, log="y") # 2)y軸をlog10で表示 boxplot(data, log="y", las=3) # 3)サンプル名を縦書きで表示 boxplot(data, log="y", las=3, # 4)ラベル情報も追加 xlab="samples", ylab="signal intensity")# 4)ラベル情報も追加 summary(data[,1:3])
p43:
summary(data[,1]) mean(data[,1], trim=0.02) apply(data, 2, mean, trim=0.02)
p44の網掛け部分:
作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提です。
hoge2.txtおよびhoge2.pngと同じものができていると思います。
{kind=link}
out_f1 <- "hoge2.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "hoge2.png" #出力ファイル名を指定してout_f2に格納 param_fig <- c(500, 300) #ファイル出力時の横幅と縦幅を指定 ### MAS5前処理法を実行し、ファイルに保存 ### library(affy) #パッケージの読み込み hoge <- ReadAffy() #*.CELファイルの読み込み eset <- mas5(hoge, normalize=F) #MAS5を実行し、結果をesetに保存 write.exprs(eset, file=out_f1) #結果をout_f1で指定したファイル名で保存。 ### 箱ひげ図を描画し、ファイルに保存 ### data <- exprs(eset) #exprs(eset)をdataに代入 colnames(data) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) png(out_f2, width=param_fig[1], height=param_fig[2]) boxplot(data, log="y", las=3, #boxplot xlab="samples", ylab="signal intensity")#boxplot dev.off() #おまじない
p44下:
list.files() apply(data, 2, mean, trim=0.02) mean(data[,1], trim=0.02) NF <- 500/mean(data[,1], trim=0.02) NF
p45:
summary(data[,1]) normalized <- data[,1]*NF summary(normalized)
書籍 | トランスクリプトーム解析 | 2.2.3 データの正規化(計算例)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp45-62のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"に移動し以下をコピペ。
p45:
作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提です。
library(affy) #パッケージの読み込み hoge <- ReadAffy() #*.CELファイルの読み込み hoge sampleNames(hoge)
p46:
hoge[1] sampleNames(hoge[1]) probesignal <- hoge[,1] sampleNames(probesignal) eset <- mas5(probesignal, normalize=F) summary(exprs(eset))
p47:
param <- "1369078_at"
exprs(eset)[param,]
PM <- pm(probesignal, param)
PM
MM <- mm(probesignal, param)
MM
p48の網掛け部分:
「以下にエラー library(param1, character.only = T) : ‘rat2302probe’ という名前のパッケージはありません」 となってしまった人は、予めrat2302probeパッケージのインストールをしておく必要がありますので、以下に新たに追加した最初の2行を実行してください。 コピペ実行後に「Update all/some/none? [a/s/n]: 」というメッセージが出ます。私はnにします。
その他のプローブ配列関連パッケージのインストールについてはイントロ | プローブ配列情報取得 | Rのパッケージからが参考になるでしょう。
hoge3.txtと同じものができていると思います。
source("http://www.bioconductor.org/biocLite.R")#書籍と違っているところ
biocLite("rat2302probe") #書籍と違っているところ
out_f <- "hoge3.txt" #出力ファイル名を指定してout_fに格納
param1 <- "rat2302probe" #パッケージ名を指定
param2 <- "1369078_at" #プローブセットID情報を指定
### 必要なパッケージをロード ###
library(Biostrings) #パッケージの読み込み
library(param1, character.only=T) #param1で指定したパッケージの読み込み
### 前処理(統一的なオブジェクト名に変更しているだけ) ###
hoge <- eval(parse(text=param1)) #オブジェクト名の変更(hogeとして取り扱う)
### 本番(フィルタリング) ###
obj <- is.element(as.character(hoge$Probe.Set.Name), param2)#条件判定
out <- hoge[obj,] #objがTRUEとなる行のみ抽出した結果をoutに格納
### 後処理(FASTA形式に変換) ###
fasta <- DNAStringSet(out$sequence) #DNAStringSetオブジェクトに変換
des <- paste(out$Probe.Set.Name,out$Probe.Interrogation.Position,sep=".")
names(fasta) <- des #description行に相当する記述を追加している
### ファイルに保存 ###
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
p48下:
out$sequence param3 <- 13 #置換したい塩基の位置を指定 ### 関数の作成 ### s_chartr <- function(str, p) { t <- substring(str, p, p) #置換したい位置の塩基を抽出 t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成 substring(str, p, p) <- t_c #置換 return(str) #置換後のデータを返す }
p49上:
MMseq <- s_chartr(out$sequence, param3)#関数を実行
MMseq
p49の網掛け部分:
fig2-3.pngと同じものができていると思います。
{kind=link}
out_f <- "fig2-3.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(500, 300) #ファイル出力時の横幅と縦幅を指定 param <- "1369078_at" #probeset IDを指定 ### シグナル強度情報取得 ### library(affy) hoge <- ReadAffy() probesignal <- hoge[,1] sampleNames(probesignal) PM <- pm(probesignal, param) MM <- mm(probesignal, param) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) min <- min(c(PM, MM)) #y軸の範囲情報(最小値)を取得 max <- max(c(PM, MM)) #y軸の範囲情報(最大値)を取得 ylim <- c(min, max) #y軸の範囲情報をylimに格納 plot(PM, ylim=ylim, ylab="Intensity", pch=20, type="o", col="black") par(new=T) #重ね書き指定 plot(MM, ylim=ylim, ylab="",pch=20, type="o", col="gray") title(param) #タイトルを追加 legend("topright", c("PM", "MM"),col=c("black", "gray"), pch=20)#凡例を追加 dev.off() #おまじない
p49下:
(671/87 + 627/297 + 969/234 + 478/197 + 770/81 + 473/97 + 258/194 + 193/286 + 194/83 + 197/58 + 232/92)/11
p50:
mean(PM/MM) mean(log2(PM/MM)) mean(log(PM/MM, base=2)) mean(log2(PM) - log2(MM))
p51:
(4 + 1/4)/2 (log2(4) + log2(1/4))/2 log2(mean(PM/MM)) # 間違い mean(log2(PM/MM)) # 正解 median(log2(PM/MM)) # 中央値 mean(log2(PM/MM), trim=0.1) # トリム平均 sort(log2(PM/MM)) # log比のソート結果を表示 sort(log2(PM/MM))[2:10] # 自力でトリム後のベクトルを作成 mean(sort(log2(PM/MM))[2:10]) # 別手段
p52:
x <- log2(PM/MM) x library(affy) tukey.biweight(x)
p53:
M <- median(x) S <- mad(x, constant=1) M S abs(x - M) median(abs(x - M))
p54:
mean(sort(log2(PM/MM))[2:10]) # 別手段 set.seed(1000) # 再現性のある乱数を発生させるためのおまじない x <- rnorm(10000000) # 標準正規分布に従う乱数を発生 sd(x) # SD mad(x, constant=1) # MAD mad(x) # MAD mad(x, constant=1.4826) # MAD
p54おまけ(図2-4を作成したad hocなコード):
fig2-4.pngと同じものができていると思います。
{kind=link}
out_f <- "fig2-4.png" # 出力ファイル名を指定してout_fに格納 param_fig <- c(400, 350) #ファイル出力時の横幅と縦幅を指定 png(out_f, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 x <- log2(PM/MM) plot(x, ylim=c(M-3*S, M+3*S), ylab="xj",# xjをプロット xlab="j", pch=19, col="black") # xjをプロット abline(h=M, col="black", lwd=1) # Mの線を引く arrows(8, M, 8, M+1*S, code=3, length=0.13)# j=8のところで上向きの矢印を描く arrows(8, M, 8, M-1*S, code=3, length=0.13)# j=8のところで上向きの矢印を描く abline(h=M+1*S, col="black", lwd=1, lty=2)# M+1*MAD abline(h=M-1*S, col="black", lwd=1, lty=2)# M-1*MAD legend("topright", "M", col="black", lty=1)# 図の凡例を追加 dev.off() # おまじない
p55:
x <- log2(PM/MM) # 書籍と違っているところ(上のおまけを実行しなかったらxオブジェクトには標準正規分布乱数が入ったままなので...) c <- 5 e <- 0.0001 u <- (x - M)/(c*S + e) u
p56:
w <- 1 - abs(u)
matome <- cbind(x, u, w)
colnames(matome) <- c("x", "u", "w")
matome
p56おまけ(図2-5を作成したad hocなコード):
fig2-5.pngと同じものができていると思います。
{kind=link}
out_f <- "fig2-5.png" # 出力ファイル名を指定してout_fに格納 param_fig <- c(400, 350) #ファイル出力時の横幅と縦幅を指定 png(out_f, width=param_fig[1], height=param_fig[2])#出力ファイルの各種パラメータを指定 plot(u, ylim=c(-1, 1), ylab="uj", # ujをプロット xlab="j", pch=19, col="black") # ujをプロット for(i in 1:10){ text(x=i, y=u[i], sprintf("%3.2f", w[i]), pos=4, cex=1.2) } abline(h=0, col="black", lwd=1) # u = 0の線を引く legend("topright", "M", col="black", lty=1)# 図の凡例を追加 dev.off() # おまじない
p57:
sum(w*x)/sum(w)
pnorm(1.96)*2 - 1
pnorm(2.58)*2 - 1
pnorm(3.37)*2 - 1
c <- 2
e <- 0.0001
u <- (x - M)/(c*S + e)
w <- 1 - abs(u)
matome <- cbind(x, u, w)
colnames(matome) <- c("x", "u", "w")
matome
p58:
u[abs(u) > 1] <- 1
w <- 1 - abs(u)
matome <- cbind(x, u, w)
colnames(matome) <- c("x", "u", "w")
matome
sum(w*x)/sum(w)
p59:
c <- 5
e <- 0.0001
u <- (x - M)/(c*S + e)
u[abs(u) > 1] <- 1
w <- (1 - (u)^2)^2
matome <- cbind(x, u, w)
colnames(matome) <- c("x", "u", "w")
matome
sum(w*x)/sum(w)
tukey.biweight(x, c=5, epsilon=0.0001)
w <- (1 - (abs(u))^3)^3
matome <- cbind(x, u, w)
colnames(matome) <- c("x", "u", "w")
matome
p60:
より頑健な...とは書いているものの、その明確な根拠は示していないですね。。。
sum(w*x)/sum(w) M <- tukey.biweight(x, c=5, epsilon=0.0001) S <- mad(x, center=M, constant=1) u <- (x - M)/(c*S + e) u[abs(u) > 1] <- 1 w <- (1 - (u)^2)^2 sum(w*x)/sum(w) library(RobLoxBioC) estimate(roblox(x, eps.lower=0.0, eps.upper=0.05, k=1))[1] estimate(roblox(x, eps.lower=0.0, eps.upper=0.05, k=3))[1]
p61:
estimate(roblox(x, eps.lower=0.0, eps.upper=0.05, k=10))[1]
SB <- tukey.biweight(log2(PM/MM))
IM <- MM
obj <- ((MM >= PM) & SB > 0.03)
IM[obj] <- 2^(-SB)*PM[obj]
matome <- cbind(PM, MM, IM)
colnames(matome) <- c("PM", "MM", "IM")
matome
p62:
ここまで、作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提です。
PV <- log2(PM - IM) SigLogVal <- tukey.biweight(PV) 2^SigLogVal
書籍 | トランスクリプトーム解析 | 2.2.4 データの正規化(その他)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp62-70のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"に移動し以下をコピペ。
p62-63にかけての網掛け部分:
作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提です。
hoge5.txtと同じものができていると思います。
out_f <- "hoge5.txt" #出力ファイル名を指定してout_fに格納 library(affy) #パッケージの読み込み hoge <- ReadAffy() #*.CELファイルの読み込み eset <- rma(hoge) #RMAを実行し、結果をesetに保存 write.exprs(eset, file=out_f) #結果をout_fで指定したファイル名で保存
p63:
dim(exprs(eset)) sampleNames(hoge) param <- 1:4 sampleNames(hoge[,param]) eset2 <- rma(hoge[,param]) dim(exprs(eset2)) head(exprs(eset)[1:3, 1:2]) head(exprs(eset2)[1:3, 1:2])
p64:
param <- 1:5 eset3 <- rma(hoge[,param]) param <- 5:9 sampleNames(hoge[,param]) eset4 <- rma(hoge[,param]) head(exprs(eset3)[1:3,4:5]) head(exprs(eset4)[1:3,1:2]) cor(exprs(eset3)[,5], exprs(eset4)[,1], method="spearman")
p65:
作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提です。
library(affy)
hoge <- ReadAffy()
newname <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep=""))
sampleNames(hoge) <- newname
hist(hoge) # 図2-6作成部分
normalized <- normalize(hoge)
hist(normalized) # 図2-7作成部分
dim(pm(hoge)) # PMシグナル強度の行数と列数を表示
p66:
head(pm(hoge), n=3) # PMシグナル強度の最初の3行分を表示 hoge_sorted <- apply(pm(hoge),2,sort) # 列ごとにソート head(hoge_sorted, n=7) # ソート後の行列の最初の7行分を表示 reference <- rowMeans(hoge_sorted) # 行ごとの平均値を算出 head(reference) hist(log2(reference), probability=T) # 図2-8作成部分
p67:
summary(pm(normalized)[,1:3]) normalized_sorted <- apply(pm(normalized),2,sort)# 列ごとにソート head(normalized_sorted, n=7) # ソート後の行列の最初の7行分を表示
p68:
param <- "1369078_at" # プローブセットIDを指定 PM <- pm(hoge, param) # PMシグナル情報取得 PM apply(PM, 1, sd) # アレイ間のばらつき mean(apply(PM, 1, sd)) # アレイ間のばらつきの平均 apply(PM, 2, sd) # プローブ間のばらつき mean(apply(PM, 2, sd)) # プローブ間のばらつきの平均
p69:
library(frma) eset <- frma(hoge)
書籍 | トランスクリプトーム解析 | 2.2.5 アノテーション情報
シリーズ Useful R 第7巻 トランスクリプトーム解析のp70-71のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"など任意のディレクトリに移動し以下をコピペ。
p70-71にかけての網掛け部分:
30MB弱のファイルのダウンロードですが実行に数分かかります。hoge_GPL1355.txtと同じものが得られます。
param1 <- "GSE30533" # GEO IDを指定 param2 <- "hoge_" # 出力ファイル名の最初の部分を指定 library(GEOquery) # パッケージの読み込み data <- getGEO(param1) # 指定したGEO IDのデータを取得 hoge <- sapply(data, annotation) # 用いられたアレイ情報(GPL ID)をhogeに格納 for(i in 1:length(hoge)){ # hogeの要素数(用いたアレイ数)分ループを回す out_f <- paste(param2, hoge[i], ".txt", sep="")# ファイル名を作成 out <- data[[i]]@featureData@data # アノテーション情報抽出 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F) }
p71の網掛け部分(下):
10秒程度で終わると思います。得られるhoge6.txtの中身はhoge_GPL1355.txtと同じはずです。
out_f <- "hoge6.txt" # 出力ファイル名を指定してout_fに格納 param <- "GPL1355" # 入手したいGEO IDを指定 library(GEOquery) # パッケージの読み込み data <- getGEO(param) # 指定したGEO IDのデータを取得 out <- data@dataTable@table # アノテーション情報抽出結果をoutに格納 write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
書籍 | トランスクリプトーム解析 | 2.3.1 RNA-seqデータ(FASTQファイル)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp71-73のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p72の網掛け部分:
作業ディレクトリがデスクトップという前提です。有線LANで数十分程度はかかりますのでご注意ください。
param <- "SRA000299" #取得したいSRA IDを指定 #必要なパッケージをロード library(SRAdb) #パッケージの読み込み sqlfile <- getSRAdbFile() #最新のSRAmetadb SQLiteファイルをダウンロード sra_con <- dbConnect(SQLite(), sqlfile)#DBMSへの接続(ファイルの読み込み) hoge <- sraConvert(param, sra_con=sra_con)#メタデータ取得 hoge #hogeの中身を表示
p72下:
作業ディレクトリがデスクトップで、デスクトップ上に"SRAmetadb.sqlite"というファイルが存在するという前提です。
hoge$run実行後の表示順(SRR002324, SRR002325, SRR002320, SRR002322, SRR002323, SRR002321)が書籍中の順番と異なっている場合もあるようです。 2016年3月17日にgetFASTQinfo部分でsra_conという引数を追加しましたが、 が、おそらくデータを取りに行っているENAのURL(http://www.ebi.ac.uk/ena/data/view/reports/sra/fastq_files/)がリンク切れ になっているようでうまく情報の取得ができなくなっているようです。 対策としては、「直接大元のサイトEuropean Nucleotide Archive (ENA)に行ってSRA000299 で検索をしてgzip圧縮FASTQファイルを1つ1つダウンロードする」になります(もちろんLinuxを使えないヒト向けのアドバイスです)。 例えばhttp://www.ebi.ac.uk/ena/data/view/SRA000299に辿り着いて、 「Fastq files (ftp)」列のところからダウンロードすることになります(2016.03.17)。
param <- "SRA000299" #取得したいSRA IDを指定 library(SRAdb) #パッケージの読み込み sqlfile <- "SRAmetadb.sqlite" #作業ディレクトリ上に存在するsqliteファイルをsqlfileとして取り扱う sra_con <- dbConnect(SQLite(), sqlfile)#DBMSへの接続(ファイルの読み込み) hoge <- sraConvert(param, sra_con=sra_con)#メタデータ取得 hoge #hogeの中身を表示 hoge$run k <- getFASTQinfo(hoge$run, sra_con) #hoge$runで指定したSRRから始まるIDのFASTQファイルサイズ情報などを表示 dim(k) colnames(k)
p73:
私が行ったときのhoge$run実行後の表示順は(SRR002324, SRR002325, SRR002320, SRR002322, SRR002323, SRR002321)でした。 この場合、SRR002324 (腎臓1.5pM)とSRR002322 (肝臓1.5pM)は1番目と4番目 の要素に相当するので、書籍中のc(2,4)ではなく c(1,4)を指定しています。 2016年3月17日にgetFASTQfile部分でsra_conという引数を追加しました(2016.03.17)。
k[,c(9,14,16,17)] getFASTQfile(hoge$run[c(1,4)], sra_con, srcType='ftp') list.files(pattern=".gz")
書籍 | トランスクリプトーム解析 | 2.3.2 リファレンス配列
シリーズ Useful R 第7巻 トランスクリプトーム解析のp73-78のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p74の網掛け部分:
2014年4月15日以降にRとBioconductorのインストールを行ったヒトは、 書籍中の記述と若干の違いがあるかもしれませんが単純にバージョンの違いによるものなので気にしなくて良い。
library(BSgenome) #パッケージの読み込み available.genomes() #利用可能なゲノムをリストアップ
p74下:
head(available.genomes()) hoge <- available.genomes(splitNameParts=T) dim(hoge) colnames(hoge) hoge$organism
p75上:
table(hoge$organism) installed.genomes()
p75の網掛け部分:
以下のコピペ実行後に「Update all/some/none? [a/s/n]: 」というメッセージが出ます。私はnにします。
param <- "BSgenome.Tgondii.ToxoDB.7.0" #インストールしたいパッケージ名を指定 source("http://bioconductor.org/biocLite.R") biocLite(param)
p75下:
installed.genomes()
p76の網掛け部分:
out_f <- "hoge7.txt" #出力ファイル名を指定してout_fに格納 param_dataset <- "hsapiens_gene_ensembl"#データセット名を指定(「listDatasets(useMart("ensembl"))」にリストアップされているものを指定可能) param_attribute <- "refseq_mrna" #配列を取得したい属性名を指定(「listAttributes(mart)」にリストアップされているものを指定可能) param_seqtype <- "cdna" #配列のタイプを指定(cdna, peptide, 3utr, 5utr, genomicのいずれかが指定可能です) library(biomaRt) #パッケージの読み込み mart <- useMart("ensembl",dataset=param_dataset)#データベース名("ensembl")とparam_datasetで指定したデータセット名を与えてmartに格納 hoge <- getBM(attributes=param_attribute, filters="", values="", mart=mart)#指定したIDリスト取得結果をhogeに格納 out <- getSequence(id=hoge, type=param_attribute,#指定したパラメータで配列取得した結果をoutに格納 seqType=param_seqtype, mart=mart)#指定したパラメータで配列取得した結果をoutに格納 exportFASTA(out, file=out_f) #FASTA形式で保存
p76下:
2014年4月15日、R ver. 3.0.3で実行したときにはdim(out)の結果が「31690 2」でした。 また、hoge実行結果も「66 3」でした。
dim(out)
library(biomaRt)
hoge <- listDatasets(useMart("ensembl"))
dim(hoge)
head(hoge)
p77上:
2014年4月15日、R ver. 3.0.3でのhoge実行結果は「1149 2」でした。
param_dataset <- "hsapiens_gene_ensembl"
mart <- useMart("ensembl",dataset=param_dataset)
hoge <- listAttributes(mart)
dim(hoge)
head(hoge)
p77の網掛け部分:
有線LANで10分程度かかりますのでご注意ください。
out_f <- "hoge8.txt" #出力ファイル名を指定してout_fに格納 param_dataset <- "cporcellus_gene_ensembl"#データセット名を指定(「listDatasets(useMart("ensembl"))」にリストアップされているものを指定可能) param_attribute <- "embl" #配列を取得したい属性名を指定(「listAttributes(mart)」にリストアップされているものを指定可能) param_seqtype <- "5utr" #配列のタイプを指定(cdna, peptide, 3utr, 5utr, genomicのいずれかが指定可能です) library(biomaRt) #パッケージの読み込み mart <- useMart("ensembl",dataset=param_dataset)#データベース名("ensembl")とparam_datasetで指定したデータセット名を与えてmartに格納 hoge <- getBM(attributes=param_attribute, filters="", values="", mart=mart)#指定したIDリスト取得結果をhogeに格納 out <- getSequence(id=hoge, type=param_attribute,#指定したパラメータで配列取得した結果をoutに格納 seqType=param_seqtype, mart=mart)#指定したパラメータで配列取得した結果をoutに格納 exportFASTA(out, file=out_f) #FASTA形式で保存
p77下:
2014年4月15日、R ver. 3.0.3でのdim(out)実行結果は「12987 2」でした。
dim(out) head(out, n=3) ?getBM
書籍 | トランスクリプトーム解析 | 2.3.3 アノテーション情報
シリーズ Useful R 第7巻 トランスクリプトーム解析のp78-80のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p78の網掛け部分:
作業ディレクトリがデスクトップという前提です。有線LANで数十分程度はかかりますのでご注意ください。 細かいところですが、書籍中では最後から2行目でちゃんとぶら下げができてないですねm(_ _)m。 2017年8月2日に、フィルタリング条件の指定のところを"with_ox_refseq_mrna"から"with_refseq_mrna"に変更しました (小松将大氏 提供情報)。
out_f <- "hoge9.txt" #出力ファイル名を指定してout_fに格納 param_dataset <- "hsapiens_gene_ensembl"#データセット名を指定 param_attribute <- c("refseq_mrna","hgnc_symbol","go_id")#属性名を指定 param_filter <- "with_refseq_mrna" #フィルタリング条件を指定 library(biomaRt) #パッケージの読み込み mart <- useMart("ensembl",dataset=param_dataset) out <- getBM(attributes=param_attribute, filters=param_filter, values=list(TRUE), mart=mart) write.table(out, out_f, sep="", append=F, quote=F, row.names=F)
p78下:
2014年4月16日、R ver. 3.0.3でのdim(out)実行結果は「450576 2」でした。
dim(out) param_dataset <- "hsapiens_gene_ensembl" param_attribute <- c("refseq_mrna","hgnc_symbol","go_id") library(biomaRt) mart <- useMart("ensembl",dataset=param_dataset)
p79:
2014年4月16日、R ver. 3.0.3でのdim(out)実行結果は「585078 2」、dim(listFilters(mart))の実行結果は「301 2」でした。 また、書籍中ではc(97,137)となっていますが、 バージョンによって頻繁に位置情報が変わるようなので、以下ではc("with_ox_refseq_mrna", "refseq_mrna") の位置情報objを自動取得するように変更しています。
out <- getBM(attributes=param_attribute, mart=mart)
dim(out)
out[1:15,]
dim(listFilters(mart))
head(listFilters(mart))
obj <- is.element(listFilters(mart)$name, c("with_ox_refseq_mrna", "refseq_mrna"))#書籍と違っているところ
listFilters(mart)[obj,] #書籍と違っているところ
param_filter <- "refseq_mrna"
p80:
2014年4月16日、R ver. 3.0.3では585078から450576となっていました。
out <- getBM(attributes=param_attribute, filters=param_filter, values=list(TRUE), mart=mart)
dim(out)
out <- getBM(attributes=param_attribute, filters=param_filter, mart=mart)
out <- getBM(attributes=param_attribute, mart=mart)
dim(out)
out <- subset(out, refseq_mrna != "")
dim(out)
out[1:10,]
out <- getBM(attributes=param_attribute, mart=mart)
dim(out)
obj <- out[,1] != ""
out <- out[obj,]
dim(out)
書籍 | トランスクリプトーム解析 | 2.3.4 マッピング(準備)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp81-85のRコードです。 ここではデスクトップ上にmappingというフォルダを作成し、そこで作業を行うという前提です。
「ファイル」−「ディレクトリの変更」でデスクトップ上のmappingに移動し以下をコピペ。
p81:
enkichikan <- function(fa, p) { #関数名や引数の作成
t <- substring(fa, p, p) #置換したい位置の塩基を取りだす
t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成
substring(fa, p, p) <- t_c #置換
return(fa) #置換後のデータを返す
}
enkichikan("CCGTACG", 2)
p81-82の網掛け部分:
サンプルデータ18とほぼ同じです。ref_genome.faと同じものができていると思います。
out_f <- "ref_genome.fa" #出力ファイル名を指定してout_fに格納 param1 <- c(48, 160, 100, 123) #配列長を指定 narabi <- c("A","C","G","T") #ACGTの並びを指定 param2 <- c(28, 22, 26, 24) #(A,C,G,Tの並びで)各塩基の存在比率を指定 param3 <- "chr" #FASTA形式ファイルのdescription部分を指定 param4 <- 3 #コピーを作成したい配列番号を指定 param5 <- c(2, 7) #コピー先配列の塩基置換したい位置を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #chr1-4の生成 set.seed(1000) #おまじない(同じ乱数になるようにするため) ACGTset <- rep(narabi, param2) #文字列ベクトルACGTsetを作成 hoge <- NULL #hogeというプレースホルダの作成 for(i in 1:length(param1)){ #length(param1)で表現される配列数分だけループを回す hoge <- c(hoge, paste(sample(ACGTset, param1[i], replace=T), collapse=""))#ACGTsetの文字型ベクトルからparam1[i]回分だけ復元抽出して得られた塩基配列をhogeに格納 } #chr5の生成と塩基置換導入 hoge <- c(hoge, hoge[param4]) #(param4)番目の配列をコピーして追加 hoge[length(param1)+1] <- enkichikan(hoge[length(param1)+1], param5[1])#塩基置換 hoge[length(param1)+1] <- enkichikan(hoge[length(param1)+1], param5[2])#塩基置換 #ファイルに保存 fasta <- DNAStringSet(hoge) #DNAStringSetオブジェクトに変更 names(fasta) <- paste(param3, 1:length(hoge), sep="")#description行を追加 writeXStringSet(fasta, file=out_f, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
p82:
list.files()
fasta
width(fasta)
names(fasta)
fasta[5]
p83:
DNAString_chartr <- function(fa, p) { #関数名や引数の作成
str_list <- as.character(fa) #文字列に変更
t <- substring(str_list, p, p) #置換したい位置の塩基を取りだす
t_c <- chartr("CGAT", "GCTA", t) #置換後の塩基を作成
substring(str_list, p, p) <- t_c #置換
fa_r <- DNAStringSet(str_list) #DNAStringSetオブジェクトに戻す
names(fa_r) <- names(fa) #description部分の情報を追加
return(fa_r) #置換後のデータを返す
}
p83の網掛け部分:
作業ディレクトリ中にref_genome.faとlist_sub3.txtが存在するという前提です。 sample_RNAseq1.faと同じものができていると思います。
in_f1 <- "ref_genome.fa" #入力ファイル名を指定してin_f1に格納(multi-FASTAファイル) in_f2 <- "list_sub3.txt" #入力ファイル名を指定してin_f2に格納(リストファイル) out_f <- "sample_RNAseq1.fa" #出力ファイル名を指定してout_fに格納 param <- 4 #最後のリードの塩基置換したい位置を指定 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f1, format="fasta")#in_f1で指定したファイルの読み込み posi <- read.table(in_f2) #in_f2で指定したファイルの読み込み #部分配列の抽出および最後のリードの塩基置換 hoge <- NULL #最終的に得る結果を格納するためのプレースホルダhogeを作成しているだけ for(i in 1:nrow(posi)){ #length(posi)回だけループを回す obj <- names(fasta) == posi[i,1] #条件を満たすかどうかを判定した結果をobjに格納 hoge <- append(hoge, subseq(fasta[obj], start=posi[i,2], end=posi[i,3]))#subseq関数を用いてobjがTRUEとなるもののみに対して、posi[i,2]とposi[i,3]で与えた範囲に対応する部分配列を抽出した結果をhogeに格納 } fasta <- hoge #hogeの中身をfastaに格納 fasta[nrow(posi)] <- DNAString_chartr(fasta[nrow(posi)], param)#指定した位置の塩基置換を実行した結果をfastaに格納 #ファイルに保存 description <- paste(posi[,1], posi[,2], posi[,3], sep="_")#行列posiの各列を"_"で結合したものをdescriptionに格納 names(fasta) <- description #description行に相当する記述を追加している writeXStringSet(fasta, file=out_f, format="fasta", width=50)#fastaの中身を指定したファイル名で保存
p83下:
dim(posi) posi
p84:
list.files() fasta
p85の網掛け部分(上):
single-endデータファイルの基本形です。mapping_single1.txtと同じものができていると思います。
out_f <- "mapping_single1.txt" #出力ファイル名を指定してout_fに格納 param_FN <- "sample_RNAseq1.fa" #マップしたいファイル名を指定 param_SN <- "hoge1" #任意のサンプル名を指定 out <- cbind(param_FN, param_SN) colnames(out) <- c("FileName", "SampleName") write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
p85の網掛け部分(中):
複数のsingle-endデータファイルの場合です。mapping_single2.txtと同じものができていると思います。
out_f <- "mapping_single2.txt" #出力ファイル名を指定してout_fに格納 param_FN <- c("sample_RNAseq1.fa", "sample_RNAseq2.fa")#マップしたいファイル名を指定 param_SN <- c("sample1", "sample2") #任意のサンプル名を指定 out <- cbind(param_FN, param_SN) colnames(out) <- c("FileName", "SampleName") write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
p85の網掛け部分(下):
paired-endデータファイルの基本形です。mapping_paired.txtと同じものができていると思います。
out_f <- "mapping_paired.txt" #出力ファイル名を指定してout_fに格納 param_FN1 <- "sample_paired1.fq.gz" param_FN2 <- "sample_paired2.fq.gz" param_SN <- "uge" out <- cbind(param_FN1, param_FN2, param_SN) colnames(out) <- c("FileName1", "FileName2", "SampleName") write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
書籍 | トランスクリプトーム解析 | 2.3.5 マッピング(本番)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp86-90のRコードです。 ここではデスクトップ上にmappingというフォルダを作成し、そこで作業を行うという前提です。
「ファイル」−「ディレクトリの変更」でデスクトップ上のmappingに移動し以下をコピペ。
p86の網掛け部分:
作業ディレクトリ中にmapping_single1.txt、ref_genome.fa、およびsample_RNAseq1.faのみ存在するという前提です。
in_f1 <- "mapping_single1.txt" #入力ファイル名を指定してin_f1に格納(RNA-seqファイル) in_f2 <- "ref_genome.fa" #入力ファイル名を指定してin_f2に格納(リファレンス配列) library(QuasR) #パッケージの読み込み out <- qAlign(in_f1, in_f2, splicedAlignment=F)#マッピングを行うqAlign関数を実行した結果をoutに格納
p86下:
list.files() out
p87上:
out@alignments[,1]
p87の網掛け部分:
R ver. 3.1.0とBioconductor ver. 2.14では、「エラー: 関数 "readGAlignments" を見つけることができませんでした」となりました。 readGAlignments関数はGenomicAlignmentsから提供されているので、 このパッケージに変更しています。
library(GenomicAlignments) #書籍と違っているところ tmpfname <- out@alignments[,1] for(i in 1:length(tmpfname)){ hoge <- readGAlignments(tmpfname[i]) hoge <- as.data.frame(hoge) tmp <- hoge[, c("seqnames","start","end")] out_f <- sub(".bam", ".bed", tmpfname[i]) write.table(tmp,out_f,sep="\t",append=F,quote=F,row.names=F,col.names=F) }
p87下:
list.files()
p88上:
tmp
p88の網掛け部分:
作業ディレクトリ中にmapping_single1.txt、ref_genome.fa、およびsample_RNAseq1.faのみ存在するという前提です。
in_f1 <- "mapping_single1.txt" #入力ファイル名を指定してin_f1に格納(RNA-seqファイル) in_f2 <- "ref_genome.fa" #入力ファイル名を指定してin_f2に格納(リファレンス配列) param <- "-m 1 --best --strata -v 0" #マッピング時のオプションを指定 library(QuasR) #パッケージの読み込み out <- qAlign(in_f1, in_f2, splicedAlignment=F, alignmentParameter=param)
p88下:
hoge <- readGAlignments(out@alignments[,1]) as.data.frame(hoge) out@alignmentParameter
p89上:
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
p89の網掛け部分:
ここではデスクトップ上にhumanというフォルダを作成し、そこで作業を行うという前提です。
「ファイル」−「ディレクトリの変更」でデスクトップ上のhumanに移動し以下をコピペ。
mapping_single3.txtと同じものができていると思います。out_f <- "mapping_single3.txt" #出力ファイル名を指定してout_fに格納 param_FN <- c("SRR002324.fastq.gz", "SRR002322.fastq.gz")#マップしたいファイル名を指定 param_SN <- c("Kidney", "Liver") #任意のサンプル名を指定 out <- cbind(param_FN, param_SN) colnames(out) <- c("FileName", "SampleName") write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
p89下:
getwd()
p90上:
list.files()
p90の網掛け部分:
作業ディレクトリ中にp89で作成したmapping_single3.txt、p73で取得した SRR002324.fastq.gzとSRR002322.fastq.gzの計3つのファイルが デスクトップ上のhumanフォルダ中に存在するという前提です。
"BSgenome.Hsapiens.UCSC.hg19"の代わりに2013年12月にリリースされたGenome Reference Consortium GRCh38 ("BSgenome.Hsapiens.NCBI.GRCh38")を指定することもできます。 ただし、2014年4月16日以降にR ver. 3.1.0とBioconductor ver. 2.14をインストールしてあればの話です。
in_f1 <- "mapping_single3.txt" #入力ファイル名を指定してin_f1に格納(RNA-seqファイル) in_f2 <- "BSgenome.Hsapiens.UCSC.hg19" #入力ファイル名を指定してin_f2に格納(リファレンス配列) param <- "-m 1 --best --strata -v 2" #マッピング時のオプションを指定 ### マッピング ### library(QuasR) library(Rsamtools) time_s <- proc.time() #計算時間計測用(マッピング開始時間の記録) out <- qAlign(in_f1, in_f2, splicedAlignment=F, alignmentParameter=param) time_e <- proc.time() #計算時間計測用(マッピング終了時間の記録) ### QCレポート ### out_f <- sub(".bam", "_QC.pdf", out@alignments[,1]) qQCReport(out, pdfFilename=out_f)
p90下:
alignmentStats(out) time_e - time_s
書籍 | トランスクリプトーム解析 | 2.3.6 カウントデータ取得
シリーズ Useful R 第7巻 トランスクリプトーム解析のp91-97のRコードです。 ここでは2.3.5に引き続いてデスクトップ上のhumanというフォルダ上で作業を行うという前提です。 いくつかコメントしてありますが、2014年4月17日にR ver. 3.1.0とBioconductor ver. 2.14をインストールしたノートPCで確認した結果です。 インストールの仕方についてはRのインストールと起動をごらんください。 いくつかの数値の違いは、Rのバージョンというのもあるでしょうし、取得先のデータベース側のバージョンにもよると思います。
「ファイル」−「ディレクトリの変更」でデスクトップ上のhumanに移動し以下をコピペ。
p91の網掛け部分(上):
library(GenomicFeatures) supportedUCSCtables()
p91の網掛け部分(中):
2014年4月18日に表示結果を確認したところ、ヒトゲノムの最新版は Genome Reference Consortium GRCh38で"hg38"として利用可能なようですね。
library(rtracklayer) ucscGenomes()
p91の網掛け部分(下):
p90の網掛け部分で"BSgenome.Hsapiens.UCSC.hg19"の代わりに2013年12月にリリースされたGenome Reference Consortium GRCh38 ("BSgenome.Hsapiens.NCBI.GRCh38")を指定した場合は、"hg19"のところを"hg38"とすべきです。 5分程度かかります。 2016年2月9日にmakeTranscriptDbFromUCSCをmakeTxDbFromUCSCに変更しました。
param1 <- "hg19" param2 <- "ensGene" library(GenomicFeatures) txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
p91下:
2014年4月18日、R ver. 3.1.0での転写物は204,940種類、exonは584,914領域となっていました。
txdb
p92:
2014年4月18日、R ver. 3.1.0でのdim(count)実行結果は「60234 3」でした。7分程度かかります。
count <- qCount(out, txdb, reportLevel="gene")
head(count)
dim(count)
p92の網掛け部分:
SRA000299_ensgene.txt(約1.5MB)と同じか似たものができていると思います。
out_f <- "SRA000299_ensgene.txt"
tmp <- cbind(rownames(count), count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p93の網掛け部分:
R ver. 3.1.0とBioconductor ver. 2.14では、「エラー: 関数 "readGAlignments" を見つけることができませんでした」となりました。 readGAlignments関数はGenomicAlignmentsから提供されているので、 このパッケージに変更しています。2分程度かかります。
library(GenomicAlignments) #書籍と違っているところ
tmpfname <- out@alignments[,1]
kidney <- readGAlignments(tmpfname[1])
liver <- readGAlignments(tmpfname[2])
merge <- c(kidney, liver)
m <- reduce(granges(merge))
p93下:
書籍中では最後の列の列名が"ngap"となっていましたが、R ver. 3.1.0とBioconductor ver. 2.14では"njunc"に変わっていますね。
head(as.data.frame(kidney)) head(as.data.frame(liver))
p94上:
dim(as.data.frame(kidney)) dim(as.data.frame(liver)) dim(as.data.frame(m)) head(as.data.frame(m))
p94の網掛け部分:
1分強かかります。
tmpsname <- out@alignments[,2]
tmp <- as.data.frame(m)
for(i in 1:length(tmpfname)){
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(tmp, count)
}
p94下:
tmpsname
p95上:
head(tmp) dim(tmp)
p95の網掛け部分:
SRA000299_granges.txt(約100MB)と同じか似たものができていると思います。3分程度かかります。
out_f <- "SRA000299_granges.txt"
tmpsname <- out@alignments[,2]
h <- as.data.frame(m)
tmp <- paste(h[,1],h[,2],h[,3],h[,4],h[,5], sep="_")
for(i in 1:length(tmpfname)){
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(tmp, count)
}
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p95下:
head(tmp) dim(tmp)
p96の網掛け部分:
書籍中のコード内では50の色が灰色になっていませんでしたm(_ _)m
p90の網掛け部分で"BSgenome.Hsapiens.UCSC.hg19"の代わりに2013年12月にリリースされたGenome Reference Consortium GRCh38 ("BSgenome.Hsapiens.NCBI.GRCh38")を指定した場合は、ここも"BSgenome.Hsapiens.NCBI.GRCh38"とすべきです。
SRA000299_seq.fa(約240MB)と同じか似たものができていると思います。2分弱かかります。
out_f <- "SRA000299_seq.fa" param <- "BSgenome.Hsapiens.UCSC.hg19" library(param, character.only=T) tmp <- ls(paste("package", param, sep=":")) genome <- eval(parse(text=tmp)) ### 配列取得およびファイル保存 ### fasta <- getSeq(genome, m) h <- as.data.frame(m) names(fasta) <- paste(h[,1],h[,2],h[,3],h[,4],h[,5], sep="_") writeXStringSet(fasta, file=out_f, format="fasta", width=50)
p96下:
head(fasta) head(names(fasta)) head(width(fasta))
p97の網掛け部分:
p96で作成したSRA000299_seq.faが存在するという前提です。 Rのバージョンなどによって得られる配列が異なるのでリンク先のファイルは参考程度です。 SRA000299_GC.txt(約165MB)と同じか似たものができていると思います。1分弱で終わります。
in_f <- "SRA000299_seq.fa" out_f <- "SRA000299_GC.txt" library(Biostrings) fasta <- readDNAStringSet(in_f, format="fasta") ### GC含量計算本番 ### hoge <- alphabetFrequency(fasta) #CG <- rowSums(hoge[,2:3]) #C,Gの総数を計算してCGに格納(2015年9月11日以前の記述) #ACGT <- rowSums(hoge[,1:4]) #A,C,G,Tの総数を計算してACGTに格納(2015年9月11日以前の記述) CG <- apply(as.matrix(hoge[,2:3]), 1, sum)#C,Gの総数を計算してCGに格納(2015年9月11日以降の記述) ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)#A,C,G,Tの総数を計算してACGTに格納(2015年9月11日以降の記述) GC_content <- CG/ACGT tmp <- cbind(names(fasta), CG, ACGT, width(fasta), GC_content) colnames(tmp) <- c("description", "CG", "ACGT", "Length", "GC_content") write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
p97下:
head(tmp)
書籍 | トランスクリプトーム解析 | 3.2.1 クラスタリング(データ変換や距離の定義など)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp99-107のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"など任意のディレクトリに移動し以下をコピペ。
p99の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提になっていますが、 p40で作成したMAS5データファイル(hoge1.txt)を置いてあるディレクトリであればどこでも構いません。
in_f <- "hoge1.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) data.dist <- as.dist(1 - cor(data, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out) # 図3-1作成部分
p99下:
head(data[,c(8, 10)])
p100:
cor(data[,8], data[,10], method="spearman") 1 - cor(data[,8], data[,10], method="spearman") cor(data[,8], data[,10], method="pearson") cor(rank(data[,8]), rank(data[,10]), method="pearson")
p101の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提になっていますが、 MAS5データファイル(hoge1.txt)を置いてあるディレクトリであればどこでも構いません。
in_f <- "hoge1.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) par(mfrow=c(1,2)) ### 最短距離法(single-linkage clustering) ### data.dist <- as.dist(1 - cor(data, method="spearman")) out <- hclust(data.dist, method = "single") plot(out) # 図3-2左作成部分 ### 最長距離法(complete-linkage clustering) ### data.dist <- as.dist(1 - cor(data, method="spearman")) out <- hclust(data.dist, method = "complete") plot(out) # 図3-2右作成部分
p101下:
summary(data[,8])
p102上:
head(data[,8]) mean(data[,8]) sum(data[,8] < mean(data[,8])) 25486/31099 qqnorm(data[,8])
p102の網掛け部分:
par(mfrow=c(1,2)) ### 対数変換前の"G2_3"データのQ-Qプロット ### qqnorm(data[,8]) # 図3-3左作成部分 ### 正規分布に従う乱数を10,000個発生させて描いたQ-Qプロット ### set.seed(1000) hoge <- rnorm(10000) qqnorm(hoge) # 図3-3右作成部分
p102下:
ks.test(hoge, "pnorm", mean=mean(hoge), sd=sd(hoge))
p103:
shapiro.test(hoge) shapiro.test(rnorm(5000)) hoge <- log2(data[,8]) ks.test(hoge, "pnorm", mean=mean(hoge), sd=sd(hoge))
p104:
par(mfrow=c(1,2)) qqnorm(hoge) # 図3-4左作成部分 hist(hoge) # 図3-4右作成部分
p105の網掛け部分(上):
書籍中では作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提になっていますが、 MAS5データファイル(hoge1.txt)を置いてあるディレクトリであればどこでも構いません。
in_f <- "hoge1.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) par(mfrow=c(1,2)) ### 対数変換前のデータ ### data.dist <- as.dist(1 - cor(data, method = "pearson")) out <- hclust(data.dist, method = "average") plot(out) # 図3-5左作成部分 ### 対数変換後のデータ ### data.dist <- as.dist(1 - cor(log2(data), method = "pearson")) out <- hclust(data.dist, method = "average") plot(out) # 図3-5右作成部分
p105の網掛け部分(下):
in_f <- "hoge1.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) par(mfrow=c(1,2)) ### マンハッタン距離 ### data.dist <- dist(t(log2(data)), method = "manhattan") out <- hclust(data.dist, method = "average") plot(out) # 図3-6左作成部分 ### ユークリッド距離 ### data.dist <- dist(t(log2(data)), method = "euclidean") out <- hclust(data.dist, method = "average") plot(out) # 図3-6右作成部分
p106-107の網掛け部分:
作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1t"という前提です。 Windowsの場合、コピペは「CTRLキーとALTキーを押しながら枠内で左クリック」でコード内を全選択できます。 GSE30533_rma.txtおよびGSE30533_rob.txtと同じものができていると思います。
out_f1 <- "GSE30533_rma.txt" out_f2 <- "GSE30533_rob.txt" library(affy) hoge <- ReadAffy() ### RMA正規化の実行および(対数変換後のデータなのでそのまま)ファイル保存 ### eset <- rma(hoge) write.exprs(eset, file=out_f1) data_rma <- exprs(eset) colnames(data_rma) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) ### RobLoxBioC正規化の実行、対数変換、およびファイル保存 ### library(RobLoxBioC) eset <- robloxbioc(hoge) exprs(eset) <- log2(exprs(eset)) write.exprs(eset, file=out_f2) data_rob <- exprs(eset) colnames(data_rob) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) ### サンプル間クラスタリング(左:RMAデータ、右:RobLoxBioCデータ) ### par(mfrow=c(1,2)) data.dist <- as.dist(1 - cor(data_rma, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out) # 図3-7左作成部分 data.dist <- as.dist(1 - cor(data_rob, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out) # 図3-7右作成部分
書籍 | トランスクリプトーム解析 | 3.2.2 実験デザイン, データ分布, 統計解析との関係
シリーズ Useful R 第7巻 トランスクリプトーム解析のp107-111のRコードです。 Windowsの場合、コピペは「CTRLキーとALTキーを押しながら枠内で左クリック」でコード内を全選択できます。
この項目では作業ディレクトリはどこでもかまいません。
p108-109の網掛け部分:
param_G1 <- 4 #G1群のサンプル数を指定 param_G2 <- 4 #G2群のサンプル数を指定 param_Ngene <- 10000 #遺伝子数を指定 ### ランダムデータの生成 ### set.seed(1000) hoge <- rnorm(param_Ngene*(param_G1+param_G2)) data <- matrix(hoge, nrow=param_Ngene, byrow=T) rownames(data) <- paste("gene", 1:param_Ngene, sep="_") colnames(data) <- c(paste("G1_", 1:param_G1, sep=""), paste("G2_", 1:param_G2, sep=""))
p109中:
dim(data) summary(data) apply(data, 2, sd)
p109の網掛け部分(下):
param_range <- c(-4.7, 4.7) param_label <- c("G1_1", "G1_2") ### 散布図の作成 ### plot(data[,1], data[,2], pch=20, cex=0.1,# 図3-8作成部分 xlim=param_range, ylim=param_range,# 図3-8作成部分 xlab=param_label[1], ylab=param_label[2])# 図3-8作成部分 grid(col="gray", lty="dotted") # 図3-8作成部分
p109下:
cor.test(data[,1],data[,2])
p110の網掛け部分:
par(mfrow=c(1,2)) data.dist <- as.dist(1 - cor(data, method = "pearson")) out <- hclust(data.dist, method = "average") plot(out) # 図3-9左作成部分 data.dist <- as.dist(1 - cor(data, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out) # 図3-9右作成部分
書籍 | トランスクリプトーム解析 | 3.2.3 多重比較問題
シリーズ Useful R 第7巻 トランスクリプトーム解析のp111-121のRコードです。 Windowsの場合、コピペは「CTRLキーとALTキーを押しながら枠内で左クリック」でコード内を全選択できます。
この項目では作業ディレクトリはどこでもかまいません。
p111の網掛け部分:
前項からの続きなので、dataオブジェクトがあるという前提です。
param_G1 <- 4 #G1群のサンプル数を指定 param_G2 <- 4 #G2群のサンプル数を指定 library(genefilter) data.cl <- c(rep(1, param_G1), rep(2, param_G2)) ### 本番(t検定) ### out <- rowttests(data, as.factor(data.cl))
p111下:
head(out, n=5)
p112:
data[1,] rank(data[1,]) t.test(data[1,data.cl==1], data[1,data.cl==2], var.equal=T)
p113:
data[1,data.cl==1] mean(data[1,data.cl==1]) mean(data[1,data.cl==2]) mean(data[1,data.cl==1]) - mean(data[1,data.cl==2]) ?rowttests p.value <- out$p.value head(p.value) sum(p.value < 0.05) sum(p.value < 0.01)
p114:
min(p.value)
obj <- p.value == min(p.value)
out[obj,]
data[obj,]
hist(p.value) # 図3-10作成部分
nrow(data)
p115:
threshold <- 0.05 Bonferroni_thres <- threshold/nrow(data) Bonferroni_thres sum(p.value < Bonferroni_thres)
p115-116の網掛け部分:
threshold <- c(0.001, 0.01, 0.03, 0.05, 0.10) res <- NULL for(i in 1:length(threshold)){ observed <- sum(p.value < threshold[i]) expected <- nrow(data)*threshold[i] FDR <- expected/observed res <- rbind(res, c(threshold[i], observed, expected, FDR)) } colnames(res) <- c("threshold", "observed", "expected", "FDR")
p116:
res head(data, n=3) data[1:1000,1:4] <- data[1:1000, 1:4] + 3 head(data, n=3)
p117上:
out <- rowttests(data, as.factor(data.cl)) p.value <- out$p.value hist(p.value) # 図3-11作成部分 threshold <- 0.05 Bonferroni_thres <- threshold/nrow(data) sum(p.value < Bonferroni_thres)
p117の網掛け部分:
threshold <- c(0.001, 0.01, 0.03, 0.05, 0.10, 0.15) res <- NULL for(i in 1:length(threshold)){ observed <- sum(p.value < threshold[i]) expected <- nrow(data)*threshold[i] FDR <- expected/observed res <- rbind(res, c(threshold[i], observed, expected, FDR)) } colnames(res) <- c("threshold", "observed", "expected", "FDR")
p117下:
res
p118上:
res[,2] - res[,3] q.value <- p.adjust(p.value, method="BH") sum(q.value < 0.1) sum(q.value < 0.04405286) sum(q.value < 0.12970169)
p118-119の網掛け部分:
書籍中ではhclust実行結果をoutオブジェクトとしていましたが、順番通りにやるとp119中の網掛け部分以降の「t検定結果のoutオブジェクト」 がhclust実行結果で上書きされてしまうことがわかりました。そのため、ここではhclust実行結果の格納先をoutからhogeに変更しています。
par(mfrow=c(1,2)) data.dist <- as.dist(1 - cor(data, method = "pearson")) hoge <- hclust(data.dist, method = "average") plot(hoge) # 図3-12左作成部分 data.dist <- as.dist(1 - cor(data, method = "spearman")) hoge <- hclust(data.dist, method = "average") plot(hoge) # 図3-12右作成部分
p119下:
sum(abs(out$dm) > 1) data.cl colnames(data) set.seed(1) posi <- sample(8) posi
p120:
data.cl[posi] out <- rowttests(data, as.factor(data.cl[posi])) sum(abs(out$dm) > 1) posi <- sample(8) posi out <- rowttests(data, as.factor(data.cl[posi])) sum(abs(out$dm) > 1) posi <- sample(8) posi out <- rowttests(data, as.factor(data.cl[posi])) sum(abs(out$dm) > 1)
書籍 | トランスクリプトーム解析 | 3.2.4 各種プロット(M-A plotや平均-分散プロットなど)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp121-129のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"など任意のディレクトリに移動し以下をコピペ。
p121の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提になっていますが、 MAS5データファイル(hoge1.txt)を置いてあるディレクトリであればどこでも構いません。
in_f <- "hoge1.txt" param_posi <- c(2, 4) data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) ### 散布図の作成 ### par(mfrow=c(1,2)) unlogged <- data[,param_posi] plot(unlogged, pch=20, cex=0.1) # 図3-13左作成部分 grid(col="gray", lty="dotted") # 図3-13左作成部分 logged <- log2(data[,param_posi]) plot(logged, pch=20, cex=0.1) # 図3-13右作成部分 grid(col="gray", lty="dotted") # 図3-13右作成部分
p122:
head(logged) M <- logged[,2] - logged[,1] A <- (logged[,2] + logged[,1])/2 plot(A, M) # 図3-14作成部分 grid(col="gray", lty="dotted") # 図3-14作成部分
p123:
ここでの作業は、R Graphics画面上に図3-14が表示されている状態で行います。 実行後に、図3-15が得られます。
obj <- (abs(M) >= 1) points(A[obj], M[obj], col="gray") sum(obj) sum(obj)/length(obj)
p124:
ここでの作業は、R Graphics画面上に図3-15が表示されている状態で行います。 実行後に、図3-16が得られます。
points(13, 4, col="black", cex=2, pch=19) points(3, 4, col="black", cex=2, pch=15)
p125:
obj <- (A < 5) sum(obj) summary(M[obj]) head(sort(abs(M[obj]), decreasing=T)) sort(abs(M[obj]), decreasing=T)[82] plot(A, M) # 図3-17作成部分 grid(col="gray", lty="dotted") # 図3-17作成部分(グリッド線を追加) points(A[obj], M[obj], col="gray") # 図3-17作成部分(A < 5に相当する部分を灰色にしている) obj2 <- abs(M) > 4.536463 # 図3-17作成部分(Mの絶対値が4.536463より大きい遺伝子の位置情報を取得) hoge <- as.logical(obj*obj2) # 図3-17作成部分(|M| > 4.536463かつA < 5を満たすものの位置情報を取得) points(A[hoge], M[hoge], col="black", pch=20)# 図3-17作成部分(黒丸をつけている)
p126:
obj <- (A > 10)
sum(obj)
sort(abs(M[obj]), decreasing=T)[45]
p126-127の網掛け部分:
p106-107で作成したRobLoxBioCデータファイル(GSE30533_rob.txt)が存在するディレクトリ上で作業を行うという前提です。
in_f <- "GSE30533_rob.txt" param_posi <- c(2, 4) data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") ### 散布図の作成 ### data <- data[,param_posi] M <- data[,2] - data[,1] A <- (data[,2] + data[,1])/2 plot(A, M) # 図3-18作成部分 grid(col="gray", lty="dotted") # 図3-18作成部分(グリッド線を追加)
p128の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提になっていますが、 MAS5データファイル(hoge1.txt)を置いてあるディレクトリであればどこでも構いません。
in_f <- "hoge1.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") par(mfrow=c(1,2)) ### G1群の平均-分散プロット(対数変換前) ### hoge <- data[,1:5] MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,# 図3-19左作成部分 xlim=c(1e-02,1e+08), ylim=c(1e-02,1e+08), col="black")# 図3-19左作成部分 grid(col="gray", lty="dotted") # 図3-19左作成部分(グリッド線を追加) abline(a=0, b=1, col="gray") # 図3-19左作成部分(y=xの灰色直線を追加) ### G1群の平均-分散プロット(対数変換後) ### hoge <- log2(data[,1:5]) MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, pch=20, cex=.1, xlim=c(-2,16), ylim=c(-2,16), col="black")# 図3-19右作成部分 grid(col="gray", lty="dotted") # 図3-19右作成部分(グリッド線を追加) abline(a=0, b=1, col="gray") # 図3-19右作成部分(y=xの灰色直線を追加)
p129の網掛け部分:
p106-107で作成したRMAデータファイル(GSE30533_rma.txt)とRobLoxBioCデータファイル(GSE30533_rob.txt)が存在するディレクトリ上で作業を行うという前提です。
in_f1 <- "GSE30533_rma.txt" in_f2 <- "GSE30533_rob.txt" par(mfrow=c(1,2)) ### G1群の平均-分散プロット(RMAデータ) ### data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") hoge <- 2^data[,1:5] MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, xlim=c(1e-02,1e+07), ylim=c(1e-02,1e+07), col="black") grid(col="gray", lty="dotted") # 図3-20左作成部分(グリッド線を追加) abline(a=0, b=1, col="gray") # 図3-20左作成部分(y=xの灰色直線を追加) ### G1群の平均-分散プロット(RobLoxBioCデータ) ### data <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="") hoge <- 2^data[,1:5] MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, xlim=c(1e-02,1e+07), ylim=c(1e-02,1e+07), col="black") grid(col="gray", lty="dotted") # 図3-20右作成部分(グリッド線を追加) abline(a=0, b=1, col="gray") # 図3-20右作成部分(y=xの灰色直線を追加)
書籍 | トランスクリプトーム解析 | 3.3.1 解析目的別留意点
シリーズ Useful R 第7巻 トランスクリプトーム解析のp129-132のRコードです。 ここでは2.3.6に引き続いてデスクトップ上のhumanというフォルダ上で作業を行うという前提です。 いくつかコメントしてありますが、2014年4月17日にR ver. 3.1.0とBioconductor ver. 2.14をインストールしたノートPCで確認した結果です。 インストールの仕方についてはRのインストールと起動をごらんください。 いくつかの数値の違いは、Rのバージョンというのもあるでしょうし、取得先のデータベース側のバージョンにもよると思います。 したがって、「得られるファイル」や「入力ファイルのリンク先のもの」と「手持ちの入力ファイル」が多少異なっていても気にする必要はありません。
「ファイル」−「ディレクトリの変更」でデスクトップ上のhumanに移動し以下をコピペ。
p130の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"human"という前提になっていますが、 p92で作成した配列長を含むカウントデータファイル(SRA000299_ensgene.txt; 約1.5MB)を置いてあるディレクトリであればどこでも構いません。
in_f <- "SRA000299_ensgene.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") ### 配列長 vs. リード数の両対数プロット ### plot(data[,1:2], log="xy", xlab="Length", ylab="Count") grid(col="gray", lty="dotted")
p130:
head(data, n=4) data <- data[data[,2] > 0, ] head(data, n=4) out <- lm(Kidney ~ width, data=log10(data)) abline(out, col="gray", lwd=2, lty="solid") lines(lowess(data, f=0.2), col="gray", lwd=2, lty="dashed")
書籍 | トランスクリプトーム解析 | 3.3.2 データの正規化(基礎編)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp132-137のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上のhumanに移動し以下をコピペ。
p132の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"human"という前提になっていますが、 p92で作成した配列長を含むカウントデータファイル(SRA000299_ensgene.txt; 約1.5MB)を置いてあるディレクトリであればどこでも構いません。
in_f <- "SRA000299_ensgene.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") ### 配列長lenとカウント情報countへの分割 ### len <- data[,1] count <- data[,-1] ### RPK補正のための遺伝子ごとの正規化係数nfの算出およびRPK正規化 ### nf <- 1000/len rpk <- sweep(count, 1, nf, "*")
p133:
head(count, n=3) head(rpk, n=3)
p133の網掛け部分:
fig3-22.pngと同じか似たものができていると思います。
{kind=link}
tmp <- cbind(len, rpk) plot(tmp[,1:2], log="xy", xlab="Length", ylab="RPK")# 図3-22作成部分 grid(col="gray", lty="dotted") # 図3-22作成部分(グリッド線を追加) tmp <- tmp[tmp[,2] > 0, ] out <- lm(Kidney ~ len, data=log10(tmp)) abline(out, col="gray", lwd=2, lty="solid") lines(lowess(tmp[,1:2], f=0.2), col="gray", lwd=2, lty="dashed")
p134上:
head(count) colSums(count)
p134の網掛け部分:
### RPM補正のためのサンプルごとの正規化係数nfの算出およびRPM正規化 ###
nf <- 1000000/colSums(count)
rpm <- sweep(count, 2, nf, "*")
p134下:
nf head(rpm)
p135上:
library(edgeR) rpm2 <- cpm(count) head(rpm2, n=3)
p135の網掛け部分:
### (3-2)式の定数を総リード数の平均で与える総リード数補正 ###
nf <- mean(colSums(count))/colSums(count)
normalized <- sweep(count, 2, nf, "*")
p135下:
nf head(normalized, n=4) colSums(normalized)
p136の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"human"という前提になっていますが、 p92で作成した配列長を含むカウントデータファイル(SRA000299_ensgene.txt; 約1.5MB)を置いてあるディレクトリであればどこでも構いません。
in_f <- "SRA000299_ensgene.txt" out_f <- "SRA000299_rpkm.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") ### 配列長lenとカウント情報countへの分割 ### len <- data[,1] count <- data[,-1] ### 遺伝子ごとの正規化係数nf_rpkの算出およびRPK正規化 ### nf_rpk <- 1000/len rpk <- sweep(count, 1, nf_rpk, "*") ### サンプルごとの正規化係数nf_rpmの算出およびrpkに対するRPM正規化 ### nf_rpm <- 1000000/colSums(count) rpkm <- sweep(rpk, 2, nf_rpm, "*") ### ファイルに保存 ### tmp <- cbind(rownames(rpkm), rpkm) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p136下:
head(rpkm, n=4)
書籍 | トランスクリプトーム解析 | 3.3.3 クラスタリング
シリーズ Useful R 第7巻 トランスクリプトーム解析のp137-145のRコードです。 ここではデスクトップ上にrecountというフォルダを作成し、そこで作業を行うという前提です。
「ファイル」−「ディレクトリの変更」でデスクトップ上のrecountに移動し以下をコピペ。
p138:
書籍中では作業ディレクトリがデスクトップ上の"recount"という前提になっていますが、 ReCountデータベース(Frazee et al., BMC Bioinformatics, 2011) から得られたbodymapのカウントデータファイル(bodymap_count_table.txt) およびラベル情報ファイル(bodymap_phenodata.txt)を置いてあるディレクトリであればどこでも構いません。
getwd() list.files()
p138の網掛け部分:
in_f <- "bodymap_count_table.txt" data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.dist <- as.dist(1 - cor(data, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out) # 図3-23作成部分
p138-139の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"recount"という前提になっていますが、 ReCountデータベース(Frazee et al., BMC Bioinformatics, 2011) から得られたbodymapのカウントデータファイル(bodymap_count_table.txt; 約3.2MB) およびラベル情報ファイル(bodymap_phenodata.txt)を置いてあるディレクトリであればどこでも構いません。
in_f1 <- "bodymap_count_table.txt" in_f2 <- "bodymap_phenodata.txt" ### ファイルの読み込み ### data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") ### dataオブジェクトの列名を変更 ### colnames(data) <- phenotype$tissue.type ### サンプル間クラスタリング ### data.dist <- as.dist(1 - cor(data, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out) # 図3-24作成部分
p139下:
log2(0) dim(data) sum(data == 0) sum(data == 0)/(52580*19)
p140上:
obj <- rowSums(data) != 0 dim(data[obj,]) sum(data[obj,] == 0) sum(data[obj,] == 0)/(13131*19)
p140の網掛け部分:
data.dist <- as.dist(1 - cor(data[obj,], method = "spearman"))
out <- hclust(data.dist, method = "average")
plot(out) # 図3-25作成部分
p140下:
hoge <- data == 0 apply(hoge, 2, sum)
p141:
hoge <- apply(data, 2, rank) apply(hoge, 2, min) hoge <- apply(data[obj,], 2, rank) apply(hoge, 2, min)
p142上:
sum(rowSums(data) == 0) sum(rowSums(data) == 1) sum(rowSums(data) == 2)
p142の網掛け部分:
### RPM補正のためのサンプルごとの正規化係数nfの算出およびRPM正規化 ### nf <- 1000000/colSums(data) rpm <- sweep(data, 2, nf, "*") ### RPM補正値の総和が1以下の遺伝子を除去(1よりも大きいものを残す) ### obj <- rowSums(data) > 1 dim(rpm[obj,])
p143:
colSums(data) nf*19
p143-144の網掛け部分:
in_f1 <- "bodymap_count_table.txt" in_f2 <- "bodymap_phenodata.txt" ### ファイルの読み込み ### data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="") phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="") ### dataオブジェクトの列名を変更 ### colnames(data) <- phenotype$tissue.type ### フィルタリング(総カウント数が0でなく、ユニークな発現パターンをもつもののみ) ### obj <- rowSums(data) != 0 hoge <- unique(data[obj,]) ### クラスタリング ### data.dist <- as.dist(1 - cor(hoge, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out)
p144下:
dim(data[obj,]) dim(unique(data[obj,])) dim(hoge) dim(unique(data)) dim(unique(rpm)) length(intersect(rownames(unique(rpm)), rownames(unique(data)))) setequal(rownames(unique(rpm)), rownames(unique(data))) length(setdiff(rownames(unique(rpm)), rownames(unique(data)))) length(union(rownames(unique(rpm)), rownames(unique(data))))
書籍 | トランスクリプトーム解析 | 3.3.4 各種プロット
シリーズ Useful R 第7巻 トランスクリプトーム解析のp145-165のRコードです。 ここではデスクトップ上にrecountというフォルダを作成し、そこで作業を行うという前提です。
「ファイル」−「ディレクトリの変更」でデスクトップ上のrecountに移動し以下をコピペ。
p145-146の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"recount"という前提になっていますが、 ReCountデータベース(Frazee et al., BMC Bioinformatics, 2011) から得られたbodymapのカウントデータファイル(bodymap_count_table.txt; 約3.2MB) を置いてあるディレクトリであればどこでも構いません。
なお、灰色の水平線はM = 0.08で限りなく0に近い値なので、見えづらくなっています。
in_f <- "bodymap_count_table.txt" ### ファイルの読み込みと列名変更 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data)[11:13] <- c("mixture1", "mixture2", "mixture3") ### RPM正規化およびlog2変換 ### nf <- 1000000/colSums(data) rpm <- sweep(data, 2, nf, "*") logged <- log(rpm, base=2) ### M-A plot ### M <- logged$mixture2 - logged$mixture1 A <- (logged$mixture2 + logged$mixture1)/2 plot(A, M) # 図3-26作成部分 grid(col="gray", lty="dotted") # 図3-26作成部分(グリッド線を追加) abline(h=median(M, na.rm=TRUE), col="gray", lwd=1)
p146下:
行列dataの11列目と12列目がmixture1とmixture2だということがわかっているという前提です。
hoge <- data[,11:12] head(hoge, n=4) obj <- (hoge[,1] == 1) & (hoge[,2] >= 1) sum(obj) head(hoge[obj,], n=3)
p147:
書籍中ではcol="gray"となっていますが、図3-27は塗りつぶし黒三角になっていますので、正しくはcol="black"ですm(_ _)m
points(A[obj], M[obj], col="black", pch=17)# 図3-27作成部分。書籍と違っているところ
head(rpm[obj,11:12])
head(log2(rpm[obj,11:12]))
head(cbind(A[obj], M[obj]))
p148:
median(M, na.rm=TRUE) 2^median(M, na.rm=TRUE) median(rpm$mixture2/rpm$mixture1, na.rm=TRUE) median(data$mixture2/data$mixture1, na.rm=TRUE) colSums(data[,11:13]) sum(data[,12])/sum(data[,11])
p149の網掛け部分:
in_f <- "bodymap_count_table.txt" ### ファイルの読み込みと列名変更 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data)[11:13] <- c("mixture1", "mixture2", "mixture3") ### log2変換 ### logged <- log(data, base=2) ### M-A plot ### M <- logged$mixture2 - logged$mixture1 A <- (logged$mixture2 + logged$mixture1)/2 plot(A, M) # 図3-28作成部分 grid(col="gray", lty="dotted") # 図3-28作成部分(グリッド線を追加) abline(h=median(M, na.rm=TRUE), col="gray", lwd=1)
p150:
median(M, na.rm=TRUE) log2(median(data$mixture2/data$mixture1, na.rm=TRUE)) log2(1.96)
p150の網掛け部分:
in_f <- "bodymap_count_table.txt" ### ファイルの読み込みおよび列名変更 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data)[11:13] <- c("mixture1", "mixture2", "mixture3") ### サブセットの抽出、G1群を1、G2群を2としたベクトルの作成 ### data <- data[,11:12] data.cl <- c(1,2) ### TCCパッケージを用いたM-A plot ### library(TCC) tcc <- new("TCC", data, data.cl) plot(tcc, normalize=T) # 図3-29作成部分
p151:
tcc normalized <- getNormalizedData(tcc) head(normalized, n=4) summary(normalized)
p152:
colSums(normalized) obj <- (normalized[,1] == 0) & (normalized[,2] > 0) head(normalized[obj,], n=4) dim(normalized[obj,]) nrow(normalized[obj,]) M <- log2(normalized[,2]) - log2(normalized[,1]) summary(M[obj]) obj1 <- normalized[,1] != 0 min(normalized[obj1,1]) normalized[!obj1,1] <- 1.426567 head(normalized[obj,], n=4)
p153:
「...必要性はない。」ではなく「...必要はない。」ですねm(_ _)m
M <- log2(normalized[,2]) - log2(normalized[,1])
summary(M[obj])
max(M[obj])
p154の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"recount"という前提になっていますが、 ReCountデータベース(Frazee et al., BMC Bioinformatics, 2011) から得られたmaqcのカウントデータファイル(maqc_count_table.txt; 約2.3MB) を置いてあるディレクトリであればどこでも構いません。
in_f <- "maqc_count_table.txt" ### ファイルの読み込みとラベル情報の作成 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.cl <- c(1,2) ### TCCパッケージを用いたM-A plot ### library(TCC) tcc <- new("TCC", data[,c(4,7)], data.cl) plot(tcc, normalize=T, median.lines=T) # 図3-30作成部分 legend("bottomright",c("G1:brain4","G2:brain7"))# 図3-30作成部分
p155の網掛け部分:
in_f <- "maqc_count_table.txt" ### ファイルの読み込みとラベル情報の作成 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.cl <- c(1,1,1,2,2,2) ### TCCパッケージを用いたM-A plot ### library(TCC) tcc <- new("TCC", data[,9:14], data.cl) plot(tcc, normalize=T, median.lines=T, ylim=c(-3.2, 3.2))# 図3-31作成部分 legend("bottomright",c("G1:UHR2-4","G2:UHR5-7"))# 図3-31作成部分
p156の網掛け部分:
in_f <- "maqc_count_table.txt" ### ファイルの読み込みとラベル情報の作成 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.cl <- c(1,1,1,2,2,2) ### TCCパッケージを用いたM-A plot ### library(TCC) tcc <- new("TCC", data[,c(1,2,3,8,9,10)], data.cl) plot(tcc, normalize=T, median.lines=T, ylim=c(-11, 11))# 図3-32作成部分 legend("bottomright",c("G1:brain1-3","G2:UHR1-3"))# 図3-32作成部分
p157の網掛け部分:
in_f <- "maqc_count_table.txt" ### ファイルの読み込みとラベル情報の作成 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") library(TCC) ### M-A plot (brain1-7 対 UHR1-7) ### tcc <- new("TCC", data, c(rep(1, 7), rep(2, 7))) plot(tcc,normalize=T,ylim=c(-11,11),xlim=c(-4,15),col="black",ann=F) ### M-A plot (brain1-3 対 brain4-6) ### par(new=T) tcc <- new("TCC", data[,1:6], c(rep(1, 3), rep(2, 3))) plot(tcc,normalize=T,ylim=c(-11,11),xlim=c(-4,15),col="gray",ann=F) ### M-A plot (UHR1-3 対 UHR4-6) ### par(new=T) tcc <- new("TCC", data[,8:13], c(rep(1, 3), rep(2, 3))) plot(tcc,normalize=T,ylim=c(-11,11),xlim=c(-4,15),col="gray") ### Legend ### legend("topright",c("b1-7 vs U1-7", "b1-3 vs b4-6", "U1-3 vs U4-6"), col=c("black", "gray", "gray"), pch=20)
p157-158の網掛け部分:
in_f <- "maqc_count_table.txt" ### ファイルの読み込みとラベル情報の変更 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data) <- c(rep("brain",7), rep("UHR",7)) ### フィルタリング(総カウント数が0でなく、ユニークな発現パターンをもつもののみ) ### obj <- rowSums(data) != 0 hoge <- unique(data[obj,]) ### サンプル間クラスタリング ### data.dist <- as.dist(1 - cor(hoge, method = "spearman")) out <- hclust(data.dist, method = "average") plot(out) # 図3-34作成部分
p157-158の網掛け部分を実行する別の手段(おまけ):
以下に示すように、TCC ver. 1.4.0以降で clusterSample関数を用いて実行することもできます。in_f <- "maqc_count_table.txt" ### ファイルの読み込みとラベル情報の変更 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") colnames(data) <- c(rep("brain",7), rep("UHR",7)) ### TCCパッケージを用いたサンプル間クラスタリング ### library(TCC) out <- clusterSample(data, dist.method="spearman", hclust.method="average", unique.pattern=TRUE) plot(out) # 図3-34作成部分
p158の網掛け部分(下):
plot関数中のcex=.1は拡大率に関するものであり、大きさを標準の0.1倍にするという意味です。
in_f <- "maqc_count_table.txt" ### ファイルの読み込み、サブセットの抽出、総リード数補正 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data <- data[,1:7] nf <- mean(colSums(data))/colSums(data) hoge <- sweep(data, 2, nf, "*") ### 平均-分散プロット ### MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,# 図3-35作成部分 xlim=c(1e-01, 1e+05), ylim=c(1e-01, 1e+05), col="black")# 図3-35作成部分 grid(col="gray", lty="dotted") # 図3-35作成部分 abline(a=0, b=1, col="gray") # 図3-35作成部分
p159:
set.seed(1000) ransuu <- rpois(n=8, lambda=100) ransuu mean(ransuu) var(ransuu) ransuu <- rpois(n=10000, lambda=100) mean(ransuu)
p160:
rpois関数実行結果の数値が若干異なっていますが、 基本的に乱数生成結果なので気にする必要はありません。
var(ransuu) head(MEAN) length(MEAN) rpois(n=7, lambda=MEAN[1]) rpois(n=7, lambda=MEAN[2]) rpois(n=7, lambda=MEAN[3])
p160-161の網掛け部分:
plot関数中のlog="xy"は、x軸y軸ともに対数でプロットするという指令です。
### 乱数行列の作成 ### simulated <- NULL for(i in 1:length(MEAN)){ simulated <- rbind(simulated, rpois(n=7, lambda=MEAN[i])) } ### 平均-分散プロット ### MEAN <- apply(simulated, 1, mean) VARIANCE <- apply(simulated, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,# 図3-36作成部分 xlim=c(1e-01, 1e+05), ylim=c(1e-01, 1e+05), col="black")# 図3-36作成部分 grid(col="gray", lty="dotted") # 図3-36作成部分 abline(a=0, b=1, col="gray") # 図3-36作成部分
p161-162の網掛け部分:
plot関数中のlog="xy"は、x軸y軸ともに対数でプロットするという指令です。図3-37です。
in_f <- "maqc_count_table.txt" ### ファイルの読み込み、総リード数補正 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") nf <- mean(colSums(data))/colSums(data) hoge <- sweep(data, 2, nf, "*") ### 平均-分散プロット(brain群とUHR群のデータを一緒にして計算) ### MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F, xlim=c(1e-01, 1e+07), ylim=c(1e-01, 1e+07), col="black") ### 平均-分散プロット(brain群のみのデータで計算) ### par(new=T) MEAN <- apply(hoge[,1:7], 1, mean) VARIANCE <- apply(hoge[,1:7], 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, xlim=c(1e-01, 1e+07), ylim=c(1e-01, 1e+07), col="gray") ### Legendなど ### grid(col="gray", lty="dotted") abline(a=0, b=1, col="gray") legend("bottomright",c("brain+UHR", "brain only"), col=c("black", "gray"), pch=20)
p162-163の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"recount"という前提になっていますが、 ReCountデータベース(Frazee et al., BMC Bioinformatics, 2011) から得られたgiladのカウントデータファイル(gilad_count_table.txt; 約1.5MB) を置いてあるディレクトリであればどこでも構いません。図3-38です。
in_f <- "gilad_count_table.txt" ### ファイルの読み込み、サブセットの抽出、総リード数補正 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data <- data[,1:3] nf <- mean(colSums(data))/colSums(data) hoge <- sweep(data, 2, nf, "*") ### 平均-分散プロット ### MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, xlim=c(1e-01, 1e+07), ylim=c(1e-01, 1e+07), col="black") grid(col="gray", lty="dotted") abline(a=0, b=1, col="gray")
p165の網掛け部分:
図3-39です。
in_f <- "gilad_count_table.txt" ### ファイルの読み込み、総リード数補正 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") nf <- mean(colSums(data))/colSums(data) hoge <- sweep(data, 2, nf, "*") ### 平均-分散プロット(male群とfemale群のデータを一緒にして計算) ### MEAN <- apply(hoge, 1, mean) VARIANCE <- apply(hoge, 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F, xlim=c(1e-01, 1e+07), ylim=c(1e-01, 1e+07), col="black") ### 平均-分散プロット(female群のみのデータで計算) ### par(new=T) MEAN <- apply(hoge[,4:6], 1, mean) VARIANCE <- apply(hoge[,4:6], 1, var) plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, xlim=c(1e-01, 1e+07), ylim=c(1e-01, 1e+07), col="gray") ### Legendなど ### grid(col="gray", lty="dotted") abline(a=0, b=1, col="gray") legend("bottomright",c("male+female", "female only"), col=c("black", "gray"), pch=20)
書籍 | トランスクリプトーム解析 | 4.2.1 2群間比較
シリーズ Useful R 第7巻 トランスクリプトーム解析のp167-173のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"など任意のディレクトリに移動し以下をコピペ。
p167-168の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提になっていますが、 MAS5データファイル(hoge1.txt)を置いてあるディレクトリであればどこでも構いません。
in_f <- "hoge1.txt" #入力ファイル名を指定してin_fに格納 out_f <- "GSE30533_MAS_DEG.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 5 #G1群のサンプル数を指定 param_G2 <- 5 #G2群のサンプル数を指定 ### ファイルの読み込み、log2変換、列名変更 ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data <- log2(data) colnames(data) <- c(paste("G1_", 1:5, sep=""), paste("G2_", 1:5, sep="")) data.cl <- c(rep(1, param_G1), rep(2, param_G2)) ### DEG検出 ### library(limma) design <- model.matrix(~ as.factor(data.cl)) fit <- lmFit(data, design) out <- eBayes(fit) p.value <- out$p.value[,ncol(design)] q.value <- p.adjust(p.value, method="BH") ranking <- rank(p.value) ### ファイルに保存 ### tmp <- cbind(rownames(data), data, p.value, q.value, ranking) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p168下:
topTable(out, coef=colnames(design)[ncol(design)], adjust="BH", number=8) head(sort(p.value)) head(sort(q.value)) sum(q.value < 0.1) sum(q.value < 0.05)
p169上:
length(q.value)
p169の網掛け部分:
mean_G1 <- apply(as.matrix(data[,data.cl==1]), 1, mean) mean_G2 <- apply(as.matrix(data[,data.cl==2]), 1, mean) M <- mean_G2 - mean_G1 A <- (mean_G1 + mean_G2)/2 plot(A, M) # 図4-1作成部分 grid(col="gray", lty="dotted") # 図4-1作成部分(グリッド線を追加) obj <- q.value < 0.1 points(A[obj], M[obj], col="gray", pch=15, cex=1.5)
p169下:
M[obj] A[obj]
p170の網掛け部分:
in_f <- "GSE30533_rob.txt" #入力ファイル名を指定してin_fに格納 param_G1 <- 5 #G1群のサンプル数を指定 param_G2 <- 5 #G2群のサンプル数を指定 param_FDR <- 0.1 #DEG検出時のFDR閾値を指定 ### ファイルの読み込み ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.cl <- c(rep(1, param_G1), rep(2, param_G2)) ### DEG検出 ### library(limma) design <- model.matrix(~ as.factor(data.cl)) fit <- lmFit(data, design) out <- eBayes(fit) hoge <- topTable(out,coef=colnames(design)[ncol(design)], adjust="BH",number= nrow(data)) ### M-A plot ### plot(hoge$AveExpr, hoge$logFC, xlab="AveExpr", ylab="logFC") grid(col="gray", lty="dotted") obj <- hoge$adj.P.Val < param_FDR points(hoge$AveExpr[obj], hoge$logFC[obj], col="gray", pch=15, cex=1.5)
p171:
sum(obj) head(hoge)
p172の網掛け部分:
AD <- hoge$logFC A <- hoge$AveExpr w <- (A - min(A))/(max(A) - min(A)) WAD <- AD * w names(WAD) <- rownames(hoge) ranking <- rank(-abs(WAD))
p172下:
head(WAD) head(ranking) w[4] w[2] AD[4] AD[2] AD[4]*w[4] AD[2]*w[2]
p173の網掛け部分:
### M-A plot ### plot(A, AD, xlab="AveExpr", ylab="logFC")# 図4-2作成部分 grid(col="gray", lty="dotted") # 図4-2作成部分(グリッド線を追加) ### limmaランキング結果上位10個を塗りつぶし灰色四角で表示 ### obj <- rank(hoge$P.Value) <= 10 points(A[obj], AD[obj], col="gray", pch=15, cex=1.5) ### WADランキング結果上位10個を塗りつぶし黒丸で表示 ### obj <- ranking <= 10 points(A[obj], AD[obj], col="black", pch=16, cex=1.2) ### Legend ### legend("bottomright", c("limma","WAD"), col=c("gray","black"), pch=c(15,16))
書籍 | トランスクリプトーム解析 | 4.2.2 他の実験デザイン(paired, multi-factor, 3群間)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp174-182のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップ上の"E-GEOD-30533.raw.1"など任意のディレクトリに移動し以下をコピペ。
p174:
G1 <- c(10.1, 7.5, 12.5, 9.3, 6.1) G2 <- c(10.8, 8.1, 13.0, 9.8, 6.8) t.test(G1, G2, paired=TRUE) t.test(G1, G2, paired=FALSE)
p175の網掛け部分:
書籍中では作業ディレクトリがデスクトップ上の"E-GEOD-30533.raw.1"という前提になっていますが、 RobLoxBioCデータファイル(GSE30533_rob.txt)を置いてあるディレクトリであればどこでも構いません。
in_f <- "GSE30533_rob.txt" #入力ファイル名を指定してin_fに格納 out_f <- "GSE30533_rob_pair_DEG.txt" #出力ファイル名を指定してout_fに格納 param <- 5 #個体数またはsib-ships数を指定 ### ファイルの読み込み ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") pair <- as.factor(rep(1:param, 2)) cl <- as.factor(c(rep(1, param), rep(2, param))) ### DEG検出 ### library(limma) design <- model.matrix(~ pair + cl) fit <- lmFit(data, design) out <- eBayes(fit) p.value <- out$p.value[,ncol(design)] q.value <- p.adjust(p.value, method="BH") ranking <- rank(p.value) ### ファイルに保存 ### tmp <- cbind(rownames(data), data, p.value, q.value, ranking) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p175下:
topTable(out, coef=colnames(design)[ncol(design)], adjust="BH", number=4)
p176:
pair cl design dim(design) colnames(design) colnames(design)[ncol(design)] head(out$p.value, n=3)
p177の網掛け部分:
in_f <- "GSE30533_rob.txt" #入力ファイル名を指定してin_fに格納 ### ファイルの読み込み ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") factor1 <- c(1,1,1,1,1,2,2,2,2,2) factor2 <- c(1,1,2,2,2,1,1,1,2,2) merge <- as.factor(paste(factor1, factor2, sep=".")) ### DEG検出 ### library(limma) design <- model.matrix(~ 0 + merge) fit <- lmFit(data, design)
p177:
merge design
p178の網掛け部分:
contrast <- makeContrasts(
analysis1 = merge1.2 - merge2.2,
analysis2 = merge1.1 - merge2.1,
analysis3 = (merge1.2 - merge2.2) - (merge1.1 - merge2.1),
levels = design)
fit2 <- contrasts.fit(fit, contrast)
out <- eBayes(fit2)
p178下:
head(out$p.value, n=3)
p179:
topTable(out, coef="analysis1", adjust="BH", number=3)
topTable(out, coef=1, adjust="BH", number=3)
topTable(out, coef=2, adjust="BH", number=3)
topTable(out, coef=3, adjust="BH", number=3)
vennDiagram(decideTests(out, adjust.method="BH", p.value=0.7))
p180の網掛け部分:
in_f <- "GSE30533_rob.txt" #入力ファイル名を指定してin_fに格納 out_f <- "GSE30533_rob_multif_DEG.txt" #出力ファイル名を指定してout_fに格納 ### ファイルの読み込み ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") factor1 <- c(rep("G1", 5), rep("G2", 5)) factor2 <- c("F","F","M","M","M", "F","F","F","M","M") merge <- as.factor(paste(factor1, factor2, sep=".")) ### DEG検出 ### library(limma) design <- model.matrix(~ 0 + merge) colnames(design) <- levels(merge) fit <- lmFit(data, design) contrast <- makeContrasts( G1vsG2inM = G1.M - G2.M, G1vsG2inF = G1.F - G2.F, Diff = (G1.M - G2.M) - (G1.F - G2.F), levels = design) fit2 <- contrasts.fit(fit, contrast) out <- eBayes(fit2) ### ファイルに保存 ### tmp <- cbind(rownames(data), data, out$p.value)
p180おまけ:
p179と同様な解析
topTable(out, coef="G1vsG2inM", adjust="BH", number=3) topTable(out, coef=1, adjust="BH", number=3) topTable(out, coef=2, adjust="BH", number=3) topTable(out, coef=3, adjust="BH", number=3) vennDiagram(decideTests(out, adjust.method="BH", p.value=0.7))
p180-181の網掛け部分:
in_f <- "GSE30533_rob.txt" #入力ファイル名を指定してin_fに格納 out_f <- "GSE30533_rob_3groups_DEG.txt"#出力ファイル名を指定してout_fに格納 param_G1 <- 4 #G1群のサンプル数を指定 param_G2 <- 3 #G2群のサンプル数を指定 param_G3 <- 3 #G3群のサンプル数を指定 ### ファイルの読み込み ### data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.cl <- c(rep("G1", param_G1), rep("G2", param_G2) , rep("G3", param_G3)) ### DEG検出 ### library(limma) design <- model.matrix(~ 0 + as.factor(data.cl)) colnames(design) <- levels(as.factor(data.cl)) fit <- lmFit(data, design) contrast <- makeContrasts( G1vsG2 = G1 - G2, G1vsG3 = G1 - G3, G2vsG3 = G2 - G3, levels = design) fit2 <- contrasts.fit(fit, contrast) out <- eBayes(fit2) ### ファイルに保存 ### tmp <- cbind(rownames(data), data, out$p.value) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p181:
dim(out$p.value) head(out$p.value, n=4) param_FDR <- 0.5 sum(p.adjust(out$p.value[,1], method="BH") < param_FDR) sum(p.adjust(out$p.value[,2], method="BH") < param_FDR) sum(p.adjust(out$p.value[,3], method="BH") < param_FDR) vennDiagram(decideTests(out, adjust.method="BH", p.value=0.5)) q.value <- apply(out$p.value, MARGIN=2, p.adjust, method="BH") head(q.value, n=4)
p182:
sum(q.value[,3] < 0.5)
書籍 | トランスクリプトーム解析 | 4.2.3 多群間比較(特異的発現パターン)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp182-188のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p182:
library(TCC)
data(hypoData_ts)
dim(hypoData_ts)
?hypoData_ts
hypoData_ts[1,]
p183の網掛け部分(上):
data <- hypoData_ts
par(mfrow=c(2,4)) #2行×4列からなる図を作成し、上の行(row)から順番に埋めていくという宣言
for(i in 1:nrow(data)){
plot(data[i,], ylab="Expression", xlab=rownames(data)[i], ylim=c(0,10))
}
p183の網掛け部分(下):
fig4-5.pngと同じものができていると思います。
{kind=link}
out_f <- "fig4-5.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(600, 350) #ファイル出力時の横幅と縦幅を指定 library(TCC) data(hypoData_ts) data <- hypoData_ts ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) par(mfrow=c(2,4)) for(i in 1:nrow(data)){ plot(data[i,], ylab="Expression", xlab=rownames(data)[i], ylim=c(0,10), cex=2.0, cex.lab=1.7, cex.axis=1.5, pch=1) } dev.off()
p184の網掛け部分:
result_ROKU.txtと同じものができていると思います。
out_f <- "result_ROKU.txt" #出力ファイル名を指定してout_fに格納 ### ファイルの読み込み ### library(TCC) data(hypoData_ts) data <- hypoData_ts ### ROKUの実行 ### out <- ROKU(data, upper.limit=0.25) ### ファイルに保存 ### tmp <- cbind(rownames(data), out$outliers, out$modH, out$rank) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p184下:
dim(out$outlier) out$outlier
p186:
out$H sort(out$H)
p187:
out$Tbw x <- data[6,] Tbw <- out$Tbw[6] y <- abs(x - Tbw) x Tbw y
p187-188の網掛け部分:
fig4-6.pngと同じものができていると思います。
{kind=link}
out_f <- "fig4-6.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(600, 350) #ファイル出力時の横幅と縦幅を指定 library(TCC) data(hypoData_ts) data <- hypoData_ts ### ROKUの実行とデータ変換 ### out <- ROKU(data, upper.limit=0.25) y <- abs(data - out$Tbw) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) par(mfrow=c(2,4)) for(i in 1:nrow(data)){ ### データ変換前 ### plot(data[i,], ylab="Expression", xlab=rownames(data)[i], ylim=c(0,10),#黒丸をプロット cex=2.0, cex.lab=1.7, cex.axis=1.5, col="black", pch=1)#黒丸をプロット(pch=1は塗りつぶさない丸に相当、cex=2.0は丸の大きさを標準の2.0倍に、cex.lab=1.7は"Expression"に相当する軸のラベルの大きさを標準の1.7倍に、など) ### Tukey biweight (Tbw)の水平線 ### abline(h=out$Tbw[i], lty="dashed", col="black") ### データ変換後 ### points(y[i,], cex=2.0, cex.lab=1.7, cex.axis=1.5, col="gray", pch=17)#pch=17が塗りつぶし三角に相当 } dev.off()
p188下:
out$modH sort(out$modH)
書籍 | トランスクリプトーム解析 | 4.3.1 シミュレーションデータ(負の二項分布)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp188-196のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p189:
「1setwd("C:/Users/kadota/Desktop/")」とsetwd関数の前に1が入っていますが間違いですm(_ _)m。 原稿校正時に混入したものと思われます。
set.seed(100)
p189-190の網掛け部分:
out_f <- "hypoData1.txt" #出力ファイル名を指定してout_fに格納 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(0.5, 0.5), replicates=c(5, 5)) data <- tcc$count ### ファイルに保存 ### tmp <- cbind(rownames(data), data) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p190:
dim(data)
head(data, n=3)
set.seed(100)
p190-191の網掛け部分:
図4-7です。
out_f <- "fig4-7.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(400, 350) #ファイル出力時の横幅と縦幅を指定 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(0.5, 0.5), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) plotFCPseudocolor(tcc) dev.off()
p191:
str(tcc$simulation) dim(tcc$simulation$DEG.foldchange) head(tcc$simulation$DEG.foldchange, n=3)
p192:
set.seed(100)
p192の網掛け部分:
図4-8です。
out_f <- "fig4-8.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(450, 350) #ファイル出力時の横幅と縦幅を指定 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(0.5, 0.5), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) plot(tcc, normalize=TRUE, median.lines=TRUE) ### Legend ### legend("bottomright",c("nonDEG","DEG(G1)","DEG(G2)"), pch=c(20,20,20), col=c("black","blue","red")) dev.off()
p193:
colSums(tcc$count) mean(colSums(tcc$count)) normalized <- getNormalizedData(tcc) colSums(normalized) data.cl <- tcc$group$group logged <- log2(normalized[1:2000,]) mean_G1 <- apply(as.matrix(logged[,data.cl==1]), 1, mean) mean_G2 <- apply(as.matrix(logged[,data.cl==2]), 1, mean) M <- mean_G2 - mean_G1 length(M) M <- M[is.finite(M)] length(M) median(M) set.seed(100)
p194の網掛け部分(上):
図4-9です。
out_f <- "fig4-9.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(400, 350) #ファイル出力時の横幅と縦幅を指定 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(1.0, 0.0), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) plotFCPseudocolor(tcc) dev.off()
p194(中):
set.seed(100)
p194の網掛け部分(下):
図4-10です。
out_f <- "fig4-10.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(450, 350) #ファイル出力時の横幅と縦幅を指定 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(1.0, 0.0), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) plot(tcc, normalize=TRUE, median.lines=TRUE) ### Legend ### legend("bottomright",c("nonDEG","DEG(G1)","DEG(G2)"), pch=c(20,20,20), col=c("black","blue","red")) dev.off()
p195:
書籍中のtcc実行結果では、Count:のところは表示させていません。
colSums(tcc$count) tcc
書籍 | トランスクリプトーム解析 | 4.3.2 データの正規化(応用編)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp196-201のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p196:
set.seed(100)
p196-197の網掛け部分:
図4-11です。
out_f <- "fig4-11.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(450, 350) #ファイル出力時の横幅と縦幅を指定 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(1.0, 0.0), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### TMM正規化 ### tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) plot(tcc, normalize=TRUE, median.lines=TRUE) ### Legend ### legend("bottomright",c("nonDEG","DEG(G1)","DEG(G2)"), pch=c(20,20,20), col=c("black","blue","red")) dev.off()
p197:
tcc
p198:
2092584 * 0.8391182 data.cl <- tcc$group$group data <- tcc$count
p198の網掛け部分:
図4-12です。
out_f <- "fig4-12.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(450, 350) #ファイル出力時の横幅と縦幅を指定 ### TMM正規化 ### library(edgeR) d <- DGEList(counts=data, group=data.cl) d <- calcNormFactors(d) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) plotSmear(d) dev.off()
p200:
set.seed(100)
p200の網掛け部分:
図4-13です。
out_f <- "fig4-13.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(450, 350) #ファイル出力時の横幅と縦幅を指定 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(1.0, 0.0), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### iDEGES/edgeR正規化 ### tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger", iteration=3, FDR=0.1, floorPDEG=0.05) ### ファイルに保存 ### png(out_f, width=param_fig[1], height=param_fig[2]) plot(tcc, normalize=TRUE, median.lines=TRUE) ### Legend ### legend("bottomright",c("nonDEG","DEG(G1)","DEG(G2)"), pch=c(20,20,20), col=c("black","blue","red")) dev.off()
書籍 | トランスクリプトーム解析 | 4.3.3 2群間比較
シリーズ Useful R 第7巻 トランスクリプトーム解析のp201-208のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p201:
set.seed(100)
p201-202の網掛け部分:
out_f <- "res_biased_TCC_DEG.txt" #出力ファイル名を指定してout_fに格納 param_FDR <- 0.05 #DEG検出時のFDR閾値を指定 ### シミュレーションデータの作成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(1.0, 0.0), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### DEG検出(iDEGES/edgeR-edgeR) ### tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger", iteration=3, FDR=0.1, floorPDEG=0.05) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR) result <- getResult(tcc, sort=FALSE) ### ファイルに保存 ### tmp <- cbind(rownames(tcc$count), tcc$count, result) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p202:
2014年4月27日、R ver. 3.1.0で実行したときには入力データが同じ場合でも若干得られる結果が変わっていました(2864個から2865個など)が、 内部的に利用しているedgeRのデフォルトオプションの変更など結果が異なりうる様々な要因が存在しますので、 あまり気にする必要はありません。
head(result) sum(result$q.value < 0.05) sum(result$estimatedDEG) head(p.adjust(result$p.value, method="BH"), n=4) tcc$count["gene_96",]
p203:
result[96,]
p203の網掛け部分:
図4-14です。p201の結果を保持しているという前提です。
out_f <- "fig4-14.png" #出力ファイル名を指定してout_fに格納 param_FDR <- 0.05 #DEG検出時のFDR閾値を指定 param_fig <- c(450, 350) #ファイル出力時の横幅と縦幅を指定 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2]) plot(tcc, FDR=param_FDR, col=c("black","gray")) ### Legend ### legend("topright", c("non-DEG",paste("DEG(FDR<", param_FDR, ")", sep="")), col=c("black", "gray"), pch=16) dev.off()
p204:
head(tcc$simulation$trueDEG) table(tcc$simulation$trueDEG) head(rank(tcc$stat$p.value))
p204の網掛け部分:
図4-15です。p201の結果を保持しているという前提です。 ROC パッケージで提供されているrocdemo.sca関数をlibrary(TCC)のみで利用できているのは、 TCCパッケージ読み込み時に内部的に用いる他のパッケージも同時に読み込んでいるからです。 これがTCCリンク先のDetailsのところの 「Depends: ..., DESeq, DESeq2, edgeR, baySeq, ROC」の意味です。
out_f <- "fig4-15.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(400, 400) #ファイル出力時の横幅と縦幅を指定 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2]) roc <- rocdemo.sca(truth = as.numeric(tcc$simulation$trueDEG != 0), data = -rank(tcc$stat$p.value)) plot(roc, xlab="1-specificity", ylab="sensitivity", cex.lab=1.4) grid(col="gray", lty="dotted") dev.off()
p205:
calcAUCValue(tcc) AUC(roc) set.seed(100)
p205-206の網掛け部分:
書籍中のコードのままでは、estimateDE関数実行部分で存在しないparam_FDRを与えているためエラーが出ます。 そのため、以下ではFDR=param_FDRというオプション部分を削除しています。 なお、該当部分は結果にはなんら影響しません。理由はFDRはDEGと判定する閾値を与えているだけであり、 ここで行っているのは閾値とは関係ない発現変動順にランキングした結果のAUC値で評価しているからです。
### シミュレーションデータ生成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 12500, PDEG = 0.32, DEG.assign=c(1.0, 0.0), DEG.foldchange=c(4, 4), replicates=c(5, 5)) ### TCC中のiDEGES/edgeR-edgeRによる解析 ### tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger", iteration=3, FDR=0.1, floorPDEG=0.05) tcc <- estimateDE(tcc, test.method="edger")#書籍と違っているところ AUC_TCC <- calcAUCValue(tcc) ### samr中のSAMseq法による解析 ### library(samr) data <- tcc$count data.cl <- tcc$group$group out <- SAMseq(data, data.cl, resp.type="Two class unpaired") p.value <- samr.pvalues.from.perms(out$samr.obj$tt, out$samr.obj$ttstar) roc <- rocdemo.sca(truth = as.numeric(tcc$simulation$trueDEG != 0), data = -rank(p.value)) AUC_SAMseq <- AUC(roc)
p206:
AUC_TCC AUC_SAMseq set.seed(100)
p206の網掛け部分(下):
out_f <- "hypoData2.txt" #出力ファイル名を指定してout_fに格納 ### シミュレーションデータ生成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.20, DEG.foldchange=c(5, 3), DEG.assign=c(0.8, 0.2), replicates=c(1, 1)) data <- tcc$count ### ファイルに保存 ### tmp <- cbind(rownames(data), data) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p206下:
dim(data) head(data, n=3)
p207上:
1行ずつ独立に実行しましょう。plotFCPseudocolor(tcc)実行時に 「 以下にエラー plot.new() : figure margins too large」 となってしまったら、R Graphics画面の横幅を広げてからもう一度トライしましょう。
plot(tcc, normalize=TRUE) plotFCPseudocolor(tcc)
p207の網掛け部分:
p206で作成したhypoData2.txtが入力です。
in_f <- "hypoData2.txt" #入力ファイル名を指定してin_fに格納 out_f <- "res_hypoData2.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 1 #G1群のサンプル数を指定 param_G2 <- 1 #G2群のサンプル数を指定 param_FDR <- 0.05 #DEG検出時のFDR閾値を指定 ### 入力ファイルの読み込み、TCCクラスオブジェクトの作成 ### library(TCC) data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.cl <- c(rep(1, param_G1), rep(2, param_G2)) tcc <- new("TCC", data, data.cl) ### DEG検出(iDEGES/DESeq-DESeq) ### tcc <- calcNormFactors(tcc, norm.method="deseq", test.method="deseq", iteration=3, FDR=0.1, floorPDEG=0.05) tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR) result <- getResult(tcc, sort=FALSE) ### ファイルに保存 ### tmp <- cbind(rownames(tcc$count), tcc$count, result) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p207下:
head(result)
p208:
sum(result$estimatedDEG == 1) sum(result$q.value < 0.05) sum(result$q.value < 0.50)
p208の網掛け部分:
p207に引き続きて行うことを前提としています。図4-16です。
out_f <- "fig4-16.png" #出力ファイル名を指定してout_fに格納 param_DEG <- 500 #上位遺伝子数を指定 param_fig <- c(450, 350) #ファイル出力時の横幅と縦幅を指定 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2]) obj <- as.numeric(tcc$stat$rank <= param_DEG) + 1 plot(tcc, col.tag=obj, col=c("black","gray")) legend("topright", c("non-DEG",paste("DEG(Top", param_DEG, ")", sep="")), col=c("black", "gray"), pch=16) dev.off()
書籍 | トランスクリプトーム解析 | 4.3.4 他の実験デザイン(3群間)
シリーズ Useful R 第7巻 トランスクリプトーム解析のp209-213のRコードです。
「ファイル」−「ディレクトリの変更」でデスクトップに移動し以下をコピペ。
p209:
set.seed(100)と網掛け部分の間に「dim(data)とhead(data, n=3)」が混入しています。 これは本来網掛け部分実行後に得られるものなので間違いですm(_ _)m
set.seed(100)
p209の網掛け部分:
図4-17です。hypoData3.txtとfig4-17.png が作成されます。
{kind=link}
out_f1 <- "hypoData3.txt" #出力ファイル名を指定してout_f1に格納 out_f2 <- "fig4-17.png" #出力ファイル名を指定してout_f2に格納 ### シミュレーションデータ生成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 10012, PDEG = 0.28, replicates=c(2,3,2), DEG.assign=c(0.6,0.15,0.25), DEG.foldchange=c(5,3,0.4)) data <- tcc$count ### ファイルに保存(カウントデータ) ### tmp <- cbind(rownames(data), data) write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F) ### ファイルに保存(pseudo-colorイメージ) ### png(out_f2, width=450, height=350) plotFCPseudocolor(tcc) dev.off()
p209下:
dim(data)
p210上:
head(data, n=3)
p210の網掛け部分:
p209で作成したhypoData3.txtが入力です。
in_f <- "hypoData3.txt" #入力ファイル名を指定してin_fに格納 out_f <- "res_hypoData3.txt" #出力ファイル名を指定してout_fに格納 param_G1 <- 2 #G1群のサンプル数を指定 param_G2 <- 3 #G2群のサンプル数を指定 param_G3 <- 2 #G3群のサンプル数を指定 param_FDR <- 0.05 #DEG検出時のFDR閾値を指定 ### 入力ファイルの読み込み、TCCクラスオブジェクトの作成 ### library(TCC) data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="") data.cl <- c(rep(1, param_G1), rep(2, param_G2) , rep(3, param_G3)) tcc <- new("TCC", data, data.cl) ### DEG検出(iDEGES/edgeR-edgeR) ### tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger", iteration=3, FDR=0.1, floorPDEG=0.05) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR) result <- getResult(tcc, sort=FALSE) ### ファイルに保存 ### tmp <- cbind(rownames(tcc$count), tcc$count, result) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
p211:
2014年4月28日、R ver. 3.1.0で実行したときには入力データが同じ場合でも若干得られる結果が変わっていましたが、 内部的に利用しているedgeRのデフォルトオプションの変更など結果が異なりうる様々な要因が存在しますので、 あまり気にする必要はありません。
head(result) sum(tcc$stat$q.value < 0.05) sum(tcc$stat$q.value < 0.10) set.seed(100)
p211-212の網掛け部分:
### シミュレーションデータ生成 ### library(TCC) tcc <- simulateReadCounts(Ngene = 10012, PDEG = 0.28, replicates=c(2,3,2), DEG.assign=c(0.6,0.15,0.25), DEG.foldchange=c(5,3,0.4)) ### TCC中のiDEGES/edgeR-edgeRによる解析 ### tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger", iteration=3, FDR=0.1, floorPDEG=0.05) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR) roc_tcc <- rocdemo.sca(truth = as.numeric(tcc$simulation$trueDEG != 0), data = -tcc$stat$rank) AUC_tcc <- calcAUCValue(tcc) ### edgeRによる解析(TCC中の関数利用) ### tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger", iteration=0, FDR=0.1, floorPDEG=0.05) tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR) roc_edger <- rocdemo.sca(truth = as.numeric(tcc$simulation$trueDEG != 0), data = -tcc$stat$rank) AUC_edger <- calcAUCValue(tcc)
p212:
AUC_tcc AUC_edger
p212の網掛け部分(下):
図4-18です。先にedgeRのROC曲線を描いたのち、TCCの結果を描いています。
out_f <- "fig4-18.png" #出力ファイル名を指定してout_fに格納 param_fig <- c(400, 400) #ファイル出力時の横幅と縦幅を指定 png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2]) plot(roc_edger, col="gray", ann=F) par(new=T) plot(roc_tcc, col="black", xlab="1-specificity", ylab="sensitivity", cex.lab=1.3) grid(col="gray", lty="dotted") legend("bottomright", c("TCC","edgeR"),col=c("black","gray"), lty="solid") dev.off()
書籍 | 日本乳酸菌学会誌 | について
(このウェブページの取扱い上、書籍としていますが学会誌です)日本乳酸菌学会誌の連載原稿を書いています。 NGSデータ解析初心者用に、各種情報収集先、Linux環境構築、Linuxコマンドなど、講習会などに出なくても十分な学習効果が得られるような情報提供を目指して執筆しています。 情報もできるだけWindows用(とMacintosh用の両方にしたいとは思っています)を作成しています。 原稿PDF、ウェブ資料を含めフリーでダウンロード可能です。本文中で触れたウェブサイトのリンク先などの情報も辿れるようにしています。 以下は主要なファイルのみリストアップしています。 ダウンロードしたPDFファイルのトップページ右上にある日付のバージョンが古い場合は、 利用しているウェブページのキャッシュに残っているのが表示されてしまう現象に遭遇してしまっています。 対策は、「一時ファイルなどのキャッシュを削除」です。
- 第1回イントロダクション(2014年07月):
- 第2回GUI環境からコマンドライン環境へ(2014年11月):
- 原稿PDF
- ウェブ資料PDF(2015.07.03版; 約2MB)
- 1. VirtualBox、および2. Extension Packのインストール手順:
- Windows用(2018.09.03版; 約1MB)
- Macintosh用(2015.11.18版; 約8MB)
- 3. 仮想マシンの作成、および4. Bio-Linux 8のisoファイルからのインストール手順:
- Windows用(2015.11.19版; 約6MB)
- Macintosh用(2015.11.19版; 約5MB)
- Bio-Linux 8のovaファイルからのインストール手順(2015/11/25追加):
- Windows用(2015.11.24版; 約2MB)
- Macintosh用(2015.11.25版; 約4MB)
- 第3回Linux環境構築からNGSデータ取得まで(2015年03月):
- 原稿PDF
- ウェブ資料PDF(オリジナル版;原稿PDFと同じく、HDD150GB、約1.3億の全リードのダウンロード。)
- Windows用(2015.07.01版; 約21MB;非推奨)
- Macintosh用(2015.04.27版; 約23MB)
- ウェブ資料PDF(軽量版;原稿PDFと異なり、HDD100GB、最初の150万リードのみに制限してダウンロード。)
- Windows用(2015.12.07版; 約20MB;推奨)
- Macintosh用
- 第4回クオリティコントロールとプログラムのインストール(2015年06月):
- 原稿PDF
- ウェブ資料PDF(オリジナル版;原稿PDFと同じく、HDD150GB、約1.3億の全リードファイルからスタート。)
- Windows用(2015.09.18版; 約24MB)
- Macintosh用
- ウェブ資料PDF(軽量版;原稿PDFと異なり、HDD100GB、最初の150万リードのみのファイルからスタート。)
- Windows用(2018.05.10版; 約21MB;推奨)
- Macintosh用
- 第5回アセンブル、マッピング、そしてQC(2015年11月):
- 第6回ゲノムアセンブリ(2016年03月):
- 原稿PDF
- ウェブ資料PDF
- Windows用(2016.06.17版; 約20MB)
- Macintosh用
- (共有フォルダ設定情報を含む)連載第3回終了時点以降のovaファイルからのインストール手順:
- Windows用(2015.12.28版; 約2MB)
- Macintosh用(2015.12.28版; 約2MB)
- 第7回ロングリードアセンブリ(2016年06月):
- 第8回アセンブリ後の解析(2016年11月):
- 第9回ゲノムアノテーションとその可視化、DDBJへの登録(2017年03月):
- 第10回DDBJへの塩基配列の登録(後編)(2017年06月):
- 第11回統合データ解析環境Galaxy(2017年11月):
- 第12回Galaxy:ヒストリーとワークフロー(2018年07月):
- 第13回RNA-seq解析(その1)(2019年03月):
- 第14回RNA-seq解析(その2)(2019年11月):
- 第15回RNA-seq解析(その3)(2020年03月):
ユーザからの要望を踏まえ、ウェブ資料を更新しました。念のためオリジナル版も残してはいますが、 基本的に軽量版をご利用ください(2015.12.07追加)。
ovaファイルを得るべく、ウェブ資料を更新しました。念のためオリジナル版も残してはいますが、 基本的に軽量版をご利用ください(2015.12.11追加)。
書籍 | 日本乳酸菌学会誌 | 第1回イントロダクション
日本乳酸菌学会誌の第1回分です。
要約:
情報収集先(講義、講習会、ウェブサイトなど):
- アグリバイオインフォマティクス教育研究プログラム
- 日本バイオインフォマティクス学会(JSBi)
- NGS現場の会
- 産総研CBRC
- HPCI人材養成プログラム
- 統合TV:Kawano et al., Brief Bioinform., 2012
- WindowsでUNIX! 1. Cygwin インストール編
- WindowsでUNIX! 2. ファイル操作編
- WindowsでUNIX! 3. ファイル操作応用編
- WindowsでUNIX! 4. ファイル操作発展編
- Perlの使い方 インストール編(Windows)
- Perlの使い方 事例編(前編)
- Perlの使い方 事例編(後編)
- イルミナ社のウェビナーシリーズ
- アメリエフ社の講習会スライド
- バイオサイエンスデータベースセンター(NBDC)
データ解析環境(Linux):
- リナックス Linux 初心者の会(ディストリビューションの選択)
- Yahoo!知恵袋(LinuxとUbuntuの違いは何ですか?)
- Ubuntuインストール by BioPapyrus
- VMware Player
- VirtualBox
- Bio-Linux:Field et al., Nat Biotechnol., 2006
- ソフトウェアリスト
- FastQC:原著論文なし
- Picard
- ABySS:Simpson et al., Genome Res., 2009
- Velvet:Zerbino and Birney, Genome Res., 2008
- Bowtie2:Langmead and Salzberg, Nat. Methods, 2012
- BWA:Li and Durbin, Bioinformatics, 2009(BWA-shortの論文)
- Cufflinks:Trapnell et al., Nat Biotechnol., 2010
- MEME:Bailey et al., Nucleic Acids Res., 2006
- MAFFT:Katoh et al., Methods Mol Biol., 2014
- T-Coffee:Magis et al., Methods Mol Biol., 2014
- WebLogo:Crooks et al., Genome Res., 2004
- sequence logos:Schneider and Stephens, Nucleic Acids Res., 1990
- Cytoscape:Shannon et al., Genome Res., 2003
- Blast2:Altschul et al., J. Mol. Biol., 1990
データ解析環境(R):
- R
- CRAN
- Bioconductor:Gentleman et al., Genome Biol., 2004
- Rのインストールと起動
- 門田幸二 著シリーズ Useful R 第7巻 トランスクリプトーム解析, 金明哲 編, 共立出版, 東京, 2014.
- R-Tips
- RjpWiki
プログラミング言語:
ウェブツール:
- DDBJ Read Annotation Pipeline:Nagasaki et al., DNA Res., 2013
- 統合TVのDDBJ Read Annotation Pipeline関連番組の一例
- Galaxy:Goecks et al., Genome Biol., 2010
- P-Galaxy:Nagasaki et al., DNA Res., 2013
- DBCLS Galaxy
- Expression Atlas:Petryszak et al., Nucleic Acids Res., 2014
- ArrayExpress:Rustici et al., Nucleic Acids Res., 2013
- 統合TVのGene Expression Atlas関連番組の一例
- Cufflinks:Trapnell et al., Nat Biotechnol., 2010
- GENSCAN:Burge et al., J Mol Biol., 1997
- 解析目的別留意点やRPKMの説明はシリーズ Useful R 第7巻 トランスクリプトーム解析のp129-137あたり
- DESeq:Anders and Huber, Genome Biol, 2010
- GSE53960 (320個のFASTQファイル):Yu et al., Nat Commun., 2014
- ReCount:Frazee et al., BMC Bioinformatics, 2011
- RefEx
- ライフサイエンス統合データベースセンター (DBCLS)
Rでゲノム解析:
- Ensembl Genomes (Zerbino et al., Nucleic Acids Res., 2018)のEnsembl Bacteria
- Lactobacillus casei 12A (Taxonomy ID: 1051650):Broadbent et al., BMC Genomics, 2012
- Lactobacillus casei 12A - Download DNA sequence - Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa.gzをダウンロード後に解凍すれば、 Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.faが得られます。
- Toplevelの概念を説明しているところ
ソフトウェアRを起動し、「ファイル」−「ディレクトリの変更」で解析したいファイル("Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa")を置いてあるディレクトリに移動し以下をコピペ。
このウェブページ中の「イントロ | NGS | 読み込み | FASTA形式 | 基本情報を取得」の5.と基本的に同じです。 2014年8月16日にN50計算のところを丁寧に書き直しました。 2015年9月11日にGC含量計算のところを入力ファイルの配列数が1つの場合にもエラーなく実行できるように変更しました。
in_f <- "Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa"#入力ファイル名を指定してin_fに格納 out_f <- "result_JSLAB1.txt" #出力ファイル名を指定してout_fに格納 #必要なパッケージをロード library(Biostrings) #パッケージの読み込み #入力ファイルの読み込み fasta <- readDNAStringSet(in_f, format="fasta")#in_fで指定したファイルの読み込み #本番(基本情報取得) Total_len <- sum(width(fasta)) #配列の「トータルの長さ」を取得 Number_of_contigs <- length(fasta) #「配列数」を取得 Average_len <- mean(width(fasta)) #配列の「平均長」を取得 Median_len <- median(width(fasta)) #配列の「中央値」を取得 Max_len <- max(width(fasta)) #配列の長さの「最大値」を取得 Min_len <- min(width(fasta)) #配列の長さの「最小値」を取得 #本番(N50情報取得) sorted <- rev(sort(width(fasta))) #長さ情報を降順にソートした結果をsortedに格納 obj <- (cumsum(sorted) >= Total_len*0.5)#条件を満たすかどうかを判定した結果をobjに格納(長い配列長のものから順番に足していってTotal_lenの50%以上かどうか) N50 <- sorted[obj][1] #objがTRUEとなる1番最初の要素のみ抽出した結果をN50に格納 #本番(GC含量情報取得) hoge <- alphabetFrequency(fasta) #A,C,G,T,..の数を配列ごとにカウントした結果をhogeに格納 #CG <- rowSums(hoge[,2:3]) #C,Gの総数を計算してCGに格納(2015年9月11日以前の記述) #ACGT <- rowSums(hoge[,1:4]) #A,C,G,Tの総数を計算してACGTに格納(2015年9月11日以前の記述) CG <- apply(as.matrix(hoge[,2:3]), 1, sum)#C,Gの総数を計算してCGに格納(2015年9月11日以降の記述) ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)#A,C,G,Tの総数を計算してACGTに格納(2015年9月11日以降の記述) GC_content <- sum(CG)/sum(ACGT) #トータルのGC含量の情報を取得 #ファイルに保存 tmp <- NULL tmp <- rbind(tmp, c("Total length (bp)", Total_len)) tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs)) tmp <- rbind(tmp, c("Average length", Average_len)) tmp <- rbind(tmp, c("Median length", Median_len)) tmp <- rbind(tmp, c("Max length", Max_len)) tmp <- rbind(tmp, c("Min length", Min_len)) tmp <- rbind(tmp, c("N50", N50)) tmp <- rbind(tmp, c("GC content", GC_content)) write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)#tmpの中身を指定したファイル名で保存
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
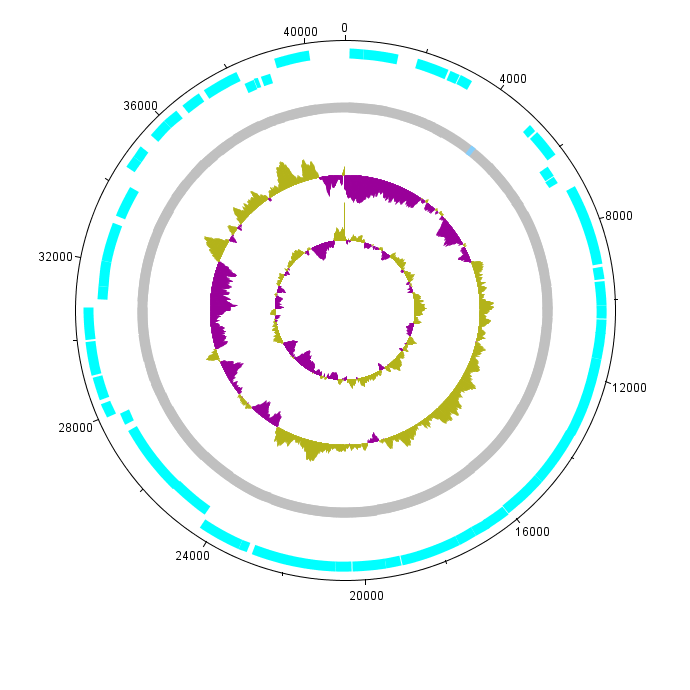{kind=link}
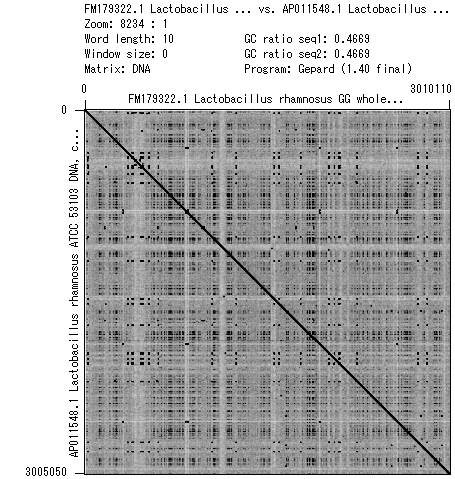{kind=link}
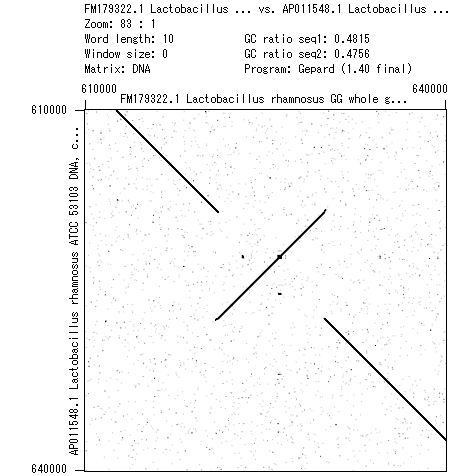{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}