(Rで)塩基配列解析
(last modified 2025/03/31, since 2010)
このページは、RStudio (R含む)で生命科学系のデータ解析を行うためのテンプレート集です。このページに特化したチュートリアル(インストール | についてと基本的な利用法)を一通り実践した上でご利用ください。より一般的なチュートリアルは、教科書の付録ページ(←読込に時間がかからなくなりました)にあるR1.010とR1.020で提供しています(2023/04/01)。
アグリバイオインフォマティクス、@Agribio_utokyo、アグリバイオの教科書、(Rで)塩基配列解析のサブページ
はじめに
このページは、主にNGS機器などから得られた塩基配列データ解析をRで行うための一連の手続きをまとめているものです。
Maintainerは東京大学・大学院農学生命科学研究科・アグリバイオインフォマティクス教育研究ユニットの門田幸二と寺田朋子です。
ボスである清水謙多郎教授をはじめ、
TCCパッケージ開発実働部隊でもあるbiopapyrus氏、
およびバグレポートや各種インストール手順書作成などで尽力いただいた諸氏のおかげでかなり規模の大きなサイトになっています
(デカくなりすぎたので、2018年7月に一部がサブページに移行しました)。
しかしながら、このサイトは2010年頃から提供しているため、(私も不具合を発見したら随時修正をしてはいますが)それでもリンク切れや内容が古いものも多々あります。
リンクも自由、講義資料などとして使うのも自由です。前もっての連絡なども必要ありません。
しかし、本当に正しいやり方かどうかなど一切の保証はできませんし、必要に応じて随時変更していますので全て自己責任でご利用ください。
間違いや修正点、また「このような解析をやりたいがどうすればいいか?」などのリクエストがあればメール(koji.kadota@gmail.com)してください。
もちろん、アグリバイオインフォマティクス教育研究プログラム受講生からのリクエストは優先的に対応します。
もし私のメアドに送ったヒトで、2勤務日以内に返事をもらってないヒトは、
アグリバイオ事務局(info あっと iu.a.u-tokyo.ac.jp)宛にも送って催促してください。
このウェブサイトは、アグリバイオインフォマティクス教育研究プログラム
はもちろんのこと、外部資金のサポートも一部受けています(過去・現在・未来)。
特に18K11521は、このウェブページの更新に特化したものです。
私のポジションがある限り、多くのユーザの効率的な研究推進の裏方として、地味~な活動を継続していければと思っております。
今後ともご支援のほど、よろしくお願いいたします。
このページ内で用いる色についての説明:
特にやらなくてもいいコマンド
プログラム実行時に目的に応じて変更すべき箇所
過去のお知らせ
- 2024年
- 生命科学研究のためのデジタルツール入門 第2版(監修:坊農秀雅・小野浩雅)が2024年6月末に出版されています。生命科学分野に参入してきた学生さんに、最新のデジタルツールを一通り学んでいただくという目的にぴったりな参考書だと思います。業界歴が長い教える側にとっては、学生さんが本書で一通りのスキルを学んでいただくことで、かなりの省力化になるのではと思います。(2024/07/11)
- 2023年
- アグリバイオインフォマティクスの教科書「Web連携テキスト バイオインフォマティクス」のページの読み込みに時間がかかる問題がありました。理由は1つの巨大なページとして構成していたためですが、章ごとのページに変更することで解決しました。(2023/05/24)
- 2023年度のアグリバイオインフォマティクス教育研究プログラムの外部生受講申し込み期間は5月9日~6月20日です。(2023/04/28)
- 「イントロ | 一般 | 配列取得 | プロモーター配列 | GenomicFeatures(Lawrence_2013)」の例題3や5を含むいくつかの場所で、シロイヌナズナのGFFやFASTAファイルのリンク切れを修正しました。(2023/04/21)
- 東京大学大学院 情報学環・学際情報学府 総合分析情報学コースの 入試説明会(2024年度4月入学、夏季入試)
が2023年4月23日(日)13:00~15:00オンラインで開催されます。(2023/04/06)
- 日本乳酸菌学会誌のNGS連載第20回の原稿を公開しました。(2023/04/01)
- 令和5年度(2023年度)もアグリバイオインフォマティクス教育研究プログラムを実施します(外部生はオンデマンド配信のみ)。多くの科目で、アグリバイオの教科書を利用した内容に順次切り替わっていきます。(2023/04/01)
- 2023年4月4日18:00-のアグリコクーン全体ガイダンスの冒頭部分で、アグリバイオインフォマティクスの簡単な紹介をさせていただきます。(2023/03/24)
- 2023年4月4日18:00-のアグリコクーン全体ガイダンスの冒頭部分で、アグリバイオインフォマティクスの簡単な紹介をさせていただきます。(2023/03/24)
- 日本乳酸菌学会誌の第19回の原稿を公開しました。(2023/01/13)
- 2022年
- 「インストール | R本体 | 過去版 | Mac用」を更新しました。(2022/12/03)
- バイオDBとウェブツール ラボで使える最新70選(小野浩雅 編)が出版されています。私がよくお世話になっているTogo picture galleryなどいろいろありますが、ざっくりと最新状況を俯瞰できてトータルで有用という位置づけだと思っています。今後も3年ごとくらいに定期的に出版されるとありがたいです。(2022/11/06)
- アグリバイオインフォマティクスの教科書「Web連携テキスト バイオインフォマティクス」が培風館より刊行されました。タイトルのWeb連携に相当する部分はこちらです。(2022/10/26)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | Blekhmanデータ | TCC(Sun_2013)」の一部の例題で挙動がおかしいようです。少なくとも例題4でエラーが出ることを私も確認済みです。この原因はTCCパッケージが内部的に用いているedgeRの仕様変更に起因します。半年ほど前まではうまく動いていたようですが、多群間比較のpost-hoc testは組み合わせも多数あるので作業が煩雑です。それゆえ、多群間比較の場合は「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+baySeq(Osabe_2019)」の例題8以降を参考にして解析するようにしてください。(2022/09/03)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+baySeq(Osabe_2019)」の例題1でエラーが出ていたので修正しました。がこの項目での推奨は例題8以降ですのでご注意ください。(2022/09/03)
- 日本乳酸菌学会誌の第18回の原稿を公開しました。(2022/08/25)
- 「実験医学別冊 論文図表を読む作法」が出版されています。タイトル通りですが、私個人としてはAccumulation curveの解説を入れていただいて大変助かっております。これまでなかなかとっつきにくかった図の理解が進む良書だと思います。(2022/07/27)
- 「正規化 | サンプル間 | 2群間 | 複製なし | iDEGES/DESeq(Sun_2013)」の項目を「推奨」としておりましたが、時代の流れでDESeqパッケージがもはや存在していないため、項目は残しつつ「推奨」という文字を削除しました。問い合わせいただいた方、どうもありがとうございました。(2022/07/27)
- 「基本的な利用法」を更新しました。特にMac版のRStudioの基本的な利用法を更新しました。(2022/06/18)
- このページをGoogle ChromeやMicrosoft Edgeで開いても、すぐにフリーズして「応答なし」と表示されることが頻繁にあったという報告をいただきました。このような事象に遭遇した方はブラウザをFirefoxに変更すると解消されるようです。情報提供いただいた学生さんに感謝m(_ _)m(2022/06/01)
- (サブページのほうのネタではありますが...)日本乳酸菌学会誌の第16回と第17回の原稿をこちらでも公開しました。(2022/05/29)
- 「インストール | R本体とRStudio | 最新版 | Mac用」を更新しました。(2022/05/16)
- 「解析 | 機能解析 | 遺伝子セット解析 | GSVA(Hänzelmann_2013)」の例題2の入力ファイルがリンク切れになっていたのを修正しました。(2022/05/13)
- 「カウント情報取得 | リアルデータ | SRP001540 | recount(Collado-Torres_2017)」の例題6と7を更新しました。(2022/05/13)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」情報を更新しました。(2022/05/13)
- 「解析 | 機能解析 | GMTファイル取得 | MSigDB(Subramanian_2005) 」を更新しました。v6.2だったのをv7.5.1に変更しました。これに関連して、v6.2のファイルを入力として読み込ませていたものも、この項目に限らず変更しました。ちゃんとチェックしきれていないので、どこかで不具合があるかもしれません。(2022/05/13)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」情報を更新しました。BSgenome.Hsapiens.NCBI.GRCh38パッケージをコメントアウトしていたのですが、機能ゲノム学の講義で使っていることが判明したので、コメントアウトを外しました。(2022/05/11)
- 「解析 | 一般 | パターンマッチング」の例題5の入力ファイル名が間違っていたのを訂正しました(data_seqlogo1.txt -> data_seqlogo1.fasta)。(中村 弘太 氏提供情報)(2022/05/09)
- 東京大学・大学院農学生命科学研究科・応用生命工学専攻の令和5(2023)年度大学院学生募集公開ガイダンスの第2回目は、5月28日(土)に開催します。(2022/05/08)
- R ver. 4.0.5でTCCパッケージのインストールがコケる現象を確認しております。この理由は、TCC内部的に利用しているDESeq2がさらに内部的に利用しているlocfitがR ver. 4.1.0以上でないといけないことに起因するようです。2022年5月1日現在の最新版はR ver. 4.2.0ですのでそれをインストールしなおすとうまくいくと期待されますので試してみてください。(2022/05/01)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC(Sun_2013)」が内部的にedgeRパッケージを用いた場合でもうまく動かなくなっていますのでご注意ください。他の「複製なし」用のスクリプトも軒並みうまく動かなくなっています。(辛川 涼眸 氏提供情報)(2022/04/29)
- 「イントロ | 一般 | 配列取得 | トランスクリプトーム配列 | biomaRt(Durinck_2009)」中のgetBM関数実行結果がベクトルから行列形式になっていたので修正しました。しかしそれでもなお、getBM関数の実行結果であるhogeオブジェクトの行数よりも、getSequence関数で得られる配列数のほうが圧倒的に多いためタイムアウトしてしまう問題は残ったままですのでご注意ください。(中村 弘太 氏提供情報)(2022/04/26)
- 「イントロ | 一般 | 指定した範囲の配列を取得 | Biostring」の例題7の入力ファイルのリンクがなくなっていたので修正しました(中村 弘太 氏提供情報)(2022/04/26)
- 東京大学・大学院農学生命科学研究科・応用生命工学専攻の令和5(2023)年度大学院学生募集公開ガイダンスは、2022年5月7日(土)と5月28日(土)に開催します。(2022/04/17)
- インストール周辺をざばっと更新しました。(2022/03/31)
- 遺伝子クラスタリングに基づく発現変動遺伝子検出法の論文(Osabe et al., BMC Bioinformatics, 2021)のプログラムをおいているMBCdegのGitHubサイトに中のコードに存在していたミスを修正しました(RNASeq.Data関数実行部分でNormalizerオプションに与える情報がlog2(size.factors)だったのをlog(size.factors)に変更)。(2022/01/22)
- 下記に関連して、「解析 | クラスタリング | RNA-seq | 遺伝子間(応用) | TCC正規化(Sun_2013)+MBCluster.Seq(Si_2014)」
の記載ミスを修正しました。RNASeq.Data関数実行部分でNormalizerオプションに与える情報はlog2(size.factors)ではなくlog(size.factors)が正しいです。下記のMBCdeg2法に相当するのが、ここの項目で提供しているコードです。(牧野 磨音 氏と私で確認;2022/01/15)
- 遺伝子クラスタリングに基づく発現変動遺伝子検出法の論文(Osabe et al., BMC Bioinformatics, 2021)のプログラムをおいているMBCdegのGitHubサイトに中のコードに一部ミスがあることが判明したので対応依頼中です。具体的には、TCC正規化係数を用いてMBCdegを実行する"MBCdeg2法"において、RNASeq.Data関数実行部分でNormalizerオプションに与える情報はlog2(size.factors)ではなくlog(size.factors)です。もちろん論文の結論には影響はありません(AUC値の分布は変わらないからです)。(牧野 磨音 氏と私で確認;2022/01/15)
- 2021年
- 2015年11月に中身を作成していた「イントロ | 一般 | 読み込み | xlsx形式 | openxlsx」という項目をリストに提示していなかったことに2021年末に気づいたので(爆)、ページ上部のリストに追加しました。(2021/12/23)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」のおそらく例題4以降でエラーが出ることを確認しておりますのでご注意ください(エラーを実際に確認したのは例題4のみ)。対応はもうちょっと先になります(ad hocな対処策を知りたいかたはお気軽にメールしてください)。すみませんm(_ _)m(平山 寛 氏提供情報;2021/12/21)
- 遺伝子クラスタリングに基づく発現変動遺伝子検出法MBCdegの論文(Osabe et al., BMC Bioinformatics, 2021)が公開されました。
コンセプト自体はDGEclust (Vavoulis et al., Genome Biol., 2015)で既に提唱されているため、
MBCdeg論文の貢献は、(1) DGEclustで提案されたコンセプトの有用性を独立して確認した、(2) DEGES正規化を組み合わせることでDGEclustの精度が上がる可能性、
そして(3) MBCdegの適用可能範囲(データ中のDEGの割合が多く偏っているような場合にはTCCよりも精度が劣る)がDGEclustにも当てはまるのではといったあたりになります。
MBCdegは、MBCluster.Seq(Si et al., Bioinformatics, 2014)をベースとしています。
MBCdeg(おそらくDGEclustも)の特徴は、我々が以前開発したRNA-seq発現変動解析用RパッケージTCC(Sun et al., BMC Bioinformatics, 2013)
よりも(試行ごとの結果のばらつきはあるものの全体としては)明らかに性能が高い点です。TCC開発者の我々が、TCCの土俵(TCCで作成したシミュレーションデータで、TCCが得意とするシナリオ)
で比較してMBCdegのほうが高い性能を示すことを確認したという点がポイントです。このあたりについては、
研究テーマの中のMBCdeg論文に関する解説のところでも触れています。(2021/10/22)
- 「作図 | M-A plot | 応用 | ggplot2編」の例題2の修正および例題3を追加しました(Manon Makino氏提供情報;2021/09/05)
- 「解析 | 一般 | オペロンDB | について」を追加しました。(2021/07/11)
- 「解析 | 一般 | オペロン | について」を「解析 | 一般 | オペロン予測 | について」に変更しました。(2021/07/12)
- 「解析 | 一般 | オペロン | について」を追加しました。(2021/07/11)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | DESeq(Anders_2010)」の項目は
DESeqパッケージが削除されているので使えなくなったことを記しました。(2021/06/01)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC(Sun_2013)」
で内部的に使っていたDESeqパッケージが削除されたのに伴い、edgeRパッケージ中の関数に切り替えました。(2021/06/01)
- 「解析 | クラスタリング | RNA-seq | 遺伝子間(応用) | TCC正規化(Sun_2013)+MBCluster.Seq(Si_2014)」
の記載ミスを修正しました。RNASeq.Data関数実行部分でNormalizerオプションに与える情報はlog(size.factors)ではなくlog2(size.factors)ですm(_ _)m(Manon Makino氏提供情報;2021/05/28)
- 「解析 | クラスタリング | RNA-seq | 遺伝子間(基礎) | MBCluster.Seq(Si_2014)」
の例題8以降の記載ミスを修正しました。RNASeq.Data関数実行部分でNormalizerオプションに与える情報はsize.factorsではなくlog2(size.factors)ですm(_ _)m(Manon Makino氏提供情報;2021/05/28)
- 「イントロ | 一般 | 配列取得 | プロモーター配列 | について」を追加しました。(2021/05/25)
- 「解析 | 一般 | CpGアイランドの同定 | について」を追加しました。(2021/05/06)
- 「イントロ | 一般 | 配列取得 | プロモーター配列 | BSgenomeとTxDbから」
の例題5の実行時にエラーが出ていることに気づいたのでそのことを記しました。(2021/04/19)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」で
SeqGSEAパッケージでエラーが出るようなので、リストから削除しました。(2021/04/02)
- 独習 Pythonバイオ情報解析が2021年3月に出版されています。一般的なプログラミング言語として解説から、塩基配列データの取り扱い、データの可視化、そしてRNA-seq解析周辺など、非常に豊富な内容となっています。編集代表の黒川顕先生にはNGSハンズオン講習会の最終年度でお世話になり、執筆者の多くの先生にはアグリバイオインフォマティクス教育研究プログラム関連講義でもお世話になっております。(2021/03/27)
- Dr.Bonoの生命科学データ解析 第2版が2021年3月に出版されています(バイオインフォマティクス初学者向けの本)。前回の第1版から3年以上経過しており、WindowsでのLinux環境(WSL2)の話など最新情報にアップデートされているのが基本形です。しかし、大枠として変わってない部分もさらっとでも読むとよいと思います。第1版当時の自分には無縁で記憶に残っていない事柄でも、今の自分と関係があるかもしれないからです(私の場合はそれがオーソログクラスターでした)。(2021/03/18)
- 令和3年度のアグリバイオインフォマティクス教育研究プログラムに関する情報をトップページに掲載しています。(2021/03/18)
- 「前処理 | クオリティコントロール | について 」をアップデートしました。(2021/01/02)
- 2020年
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | edgeR(Robinson_2010)」を更新しました。前回の最終更新が2014/07/24で、その当時と今では使われている関数も異なっているので、edgeR (ver. 3.30.3)中のUser's Guide中の1.4 Quick startに準拠しています。但し、このウェブページ全体条件を揃えるために「低発現遺伝子のフィルタリング」部分のみコメントアウトしておりますのでご注意ください。基本的に2020/10/29に注意喚起したものをやっと反映させた、という位置づけです。(2020/12/18)
- 「解析 | 機能解析 | 遺伝子セット解析 | GSVA(Hänzelmann_2013)」を更新しました。例題2のgetGmt関数内のオプションをgeneIdType=EntrezIdentifierからgeneIdType=SymbolIdentifierへと修正しました。実際問題としてはうまく動くようですが、こちらのほうが正解です(矢追 毅 氏提供情報)。(2020/11/10)
- 「解析 | 新規転写物同定(ゲノム配列を利用)」を更新しました。(2020/11/04)
- 「アセンブル | ゲノム用」を更新しました。(2020/11/04)
- 「アセンブル | トランスクリプトーム(転写物)用」を更新しました。(2020/11/03)
- 「解析 | 発現変動 | ...」の記載内容がだいぶ古くなっているのでご注意ください。
例えば、edgeRパッケージを用いてDEG検出を行う関数(例:estimateTagwiseDisp)は2015年頃の手順ですが、
今(edgeR ver. 3.30.3)はestimateDisp関数に切り替わっています。内部的にedgeRの関数を用いているTCC(ver. 1.28.0)も古いままです。
もちろん古いだけなので間違いではないですが、最近Ichihashi et al., Plant Cell Physiol., 2018の植物RNA-seqカウントデータ(2群間比較)で5% FDR (i.e., q-value < 0.05)を満たす遺伝子数を比較した際に、最新の手順で2,200個程度検出された一方、昔の手順では1,800個弱という結構な違いを目の当たりにしました。バージョンの違いでここまでの違いを見たのは初めてでしたので私自身衝撃でした。今回の結果を得るに至ったRスクリプトを20201029_TCC.txtにまとめていますので、気になった方はご確認ください。尚、TCCは次期リリース(2021年4月頃)で内部的に用いる関数を修正予定です。
尚、申し訳ありませんが、まだこのページ中のスクリプトは修正できておりませんm(_ _)m。(2020/10/29)
- 「カウント情報取得 | シミュレーションデータ | RNA-seq | について」
を更新しました。(2020/10/24)
- 東京大学大学院 情報学環・学際情報学府 総合分析情報学コースの 入試説明会(2021年度冬季入試)
が2020年10月23日(金) 18:00-20:00オンラインで開催されます。(2020/10/16)
- TCC-GUI(に限らずですが)で利用するパッケージのインストール時に書き込み権限がない的なエラーが出る場合は、
管理者権限で実行してください。また、実行時に入力データを遺伝子名(gene name)でいれると大抵の場合、
Error in data.frame: duplicate row.namesのようなエラーメッセージが出ます。
原因は同じ遺伝子名の行が複数個存在するためです。この解決策として、
例えば全ての行でユニークな文字列からなるEnsembl gene IDの情報などをお持ちでしたら、そちらをご利用ください(加藤真吾 氏提供情報)。(2020/09/22)
- 「解析 | 前処理 | フィルタリング | 低発現遺伝子 | 基礎」
で例題3を追加しました。(2020/09/22)
- 「解析 | 前処理 | フィルタリング | 低発現遺伝子 | TCC(Sun_2013)」
の記載事項が変だったので修正しました。(2020/09/22)
- R2年度はもうキャパオーバーですので、ご新規様の講演や執筆依頼はお控えくださいますようお願い申し上げます。処理能力が低くすみません。(2020/09/19)
- 2020年11月14-15日(土日)に数理生物学セミナー2020@TMDUというオンラインセミナーが開催されます。
興味ある方はどうぞ。(2020/09/19)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」で
ffbaseパッケージのインストール法を(BiocManager::installからinstall_githubへ)変更しました。
また、vcfRがCRANから削除されたので、リストから削除しました。(2020/08/25)
- 「カウント情報取得 | リアルデータ | SRP056146 | recount(Collado-Torres_2017)」
中の記載ミス(計12サンプル --> 計174サンプル)を修正しました。(2020/08/07)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+baySeq(Osabe_2019)」
を例として連絡を受けましたが、baySeqなどを実行する際に入力ファイルに実数が含まれるとうまく動きませんのでご注意ください。
例えば、STAR-RSEMで作成したものでexpected_countをそのまま入力として与えたい場合は、
入力ファイル読み込み後のオブジェクトに対してround関数を実行するなどすればよいです(茂木朋貴 氏提供情報)(2020/07/29)
- single-cell RNA-seq (scRNA-seq)の解析パイプラインのガイドラインに関する論文であるVieth et al., Nat Commun., 2019についての批評論文が公開されました
(Kadota and Shimizu, Front Genet., 2020)。特にscRNA-seqをbulk RNA-seqと差別化する際の論法や、
比較対象として用いたbulk RNA-seq用の正規化法の選定に関して、論文調査不足・事実誤認・ミスリード・不誠実さといった観点で痛烈に批判しています。(2020/07/28)
- 「解析 | クラスタリング | scRNA-seq | サンプル間 | ...」の項目でlibrary.size.normalize関数の実行に必要な
library(phateR)コマンドの追加を失念しておりましたので追加しました(山口浩史 氏提供情報)。(2020/06/04)
- 「正規化 | scRNA-seq | について」を更新しました。(2020/05/31)
- 「解析 | クラスタリング | RNA-seq | について」を更新しました。(2020/05/30)
- 日本乳酸菌学会誌のNGS関連連載の第15回分原稿PDFを公開しました。
ウェブ資料も公開しました。詳しくはサブページの「書籍 | 日本乳酸菌学会誌 | 第15回RNA-seq解析(その3)」をご覧ください。(2020/05/21)
- 「解析 | クラスタリング | scRNA-seq | サンプル間 | ...」の例題3と5を更新しました。
これまでは最適なクラスター数に関する議論まで意識してcclustパッケージのcclust関数を利用していましたが、
結果が安定しないことと安定させるためのオプションを与える術がないので、
「バイオスタティスティクス基礎論」との整合性も鑑みてstatsパッケージのkmeans関数に切り替えました。(2020/05/12)
- 「正規化 | サンプル間 | について」を更新しました。
Zhao et al., RNA, 2020の論文をリストに追加しただけですが、
私がなぜTPM (含RPKM)をほとんど教えないかの理由について同じ考えをもっていらっしゃる方の論文です。
まだpublishされていませんが(会員の方には配布済みかもしれませんが)、日本乳酸菌学会誌NGS連載第15回の原稿中にもほぼ同じことが書かれています。(2020/05/08)
- 「解析 | クラスタリング | scRNA-seq | サンプル間 | ...」あたりをいくつか更新しました。
例題4と5と追加したのがメインですが、cclust関数(k-means clustering)のパラメータチューニングが甘いのでなんか変な結果になるときがあります。(2020/05/06)
- 「解析 | クラスタリング | scRNA-seq | サンプル間 | ...」あたりをいくつか追加しました。(2020/04/21)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」の最後のパッケージインストール確認のところで、
インストールをやめたのに「library(tabplot)」で確認しようとしてエラーが出ていたのを修正しました。(2020/04/04)
- 諸般(主にコロナに対する東大の全体方針)の事情により、2020年のアグリバイオインフォマティクス教育研究プログラムは、
東京大学の学生に限定することとなりました(2020年3月17日決定)。既に応募いただいた方、そしてこれから応募しようと思っていた方々には残念なお知らせとなってしまいましたが、
ご理解いただけますと幸いですm(_ _)m (2020/03/17)
- 「作図 | 生存曲線 | 基礎」の項目をいくつか追加しました。(2020/02/23)
- 2019年
- 日本乳酸菌学会誌のNGS関連連載の第14回分原稿PDFを公開しました。ウェブ資料も公開しました。(2019/12/23)
- 「RNA-Seqデータ解析 WETラボのための鉄板レシピ(編:坊農秀雅)」が出版されています。(2019/12/23)
- TCC-GUI (Su et al., BMC Res. Notes, 2019)
の解説動画が統合TVで公開されました。DBCLSの小野さんはじめ関係者の皆様のご尽力に深謝m(_ _)m(2019/11/08)
- インストール | についての推奨手順をとりあえずWindows版(R_install_win.pdf)
のみですがアップデートし、RStudioを利用するやり方に変更しました。(2019/10/09)
- 「インストール | R本体 | 最新版 | Win用」の項目名を
「インストール | R本体とRStudio | 最新版 | Win用」に変更しました。
Mac用についても同様です。(2019/10/08)
- 「解析 | 遺伝子制御ネットワーク推定 | について」を追加しました。(2019/10/04)
- 「生命科学者のためのDr.Bonoデータ解析実践道場(著:坊農秀雅)」が出版されています。
今回の"Bono本"は、アグリバイオの大学院講義で丁寧に教えることが現実的に難しいLinux環境でのデータ解析の情報が丁寧に解説されています(アグリバイオの内容と相補的な関係)。
「聞いたことはあるがよく知らない事柄」が簡潔かつ丁寧に書かれているので、私は主にそのあたりの頭の整理に利用させてもらっています。(2019/09/30)
- 「解析 | リガンド-レセプター解析(ligand-receptor analysis) | について」を追加しました。CellPhoneDBなどを含むカテゴリです。(2019/10/04)
- 「解析 | クラスタリング | scRNA-seq | 参照情報あり | について」を追加しました。
scmapやGarnettなどを含むカテゴリです。(2019/10/03)
- 「解析 | 発現変動 | について」だった項目名を「解析 | 発現変動 | RNA-seq | について」に変更しました。
また、「解析 | 発現変動 | scRNA-seq | について」を追加しました。(2019/10/01)
- 「正規化 | scRNA-seq | について」を追加しました。(2019/09/27)
- 「解析 | ゲノム | 領域の一致の評価 | regioneR(Gel_2016)」をとりあえず項目だけ追加しました。(2019/09/27)
- 「解析 | 前処理 | scRNA-seq | についての内容を追加しました。(2019/09/26)
- 「解析 | 機械学習(分類) | 基礎 | MLSeq(Goksuluk_2019)」が一通り完成しました。(2019/09/23)
- 「解析 | 機械学習(分類) | 基礎 | MLSeq(Goksuluk_2019)」を書き進めています。(2019/09/20)
- rbamtoolsパッケージが削除されていることが判明しましたので、
「インストール | Rパッケージ | 必要最小限プラスアルファ」から消しました。(2019/09/12)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」中で、
任意の行の部分を選択できない現象が起こっているようです。デフォルトで表示されている部分より下のほうで挙動がおかしくなっています。
どなたか解決策がお分かりのかたは教えていただければ幸いですm(_ _)m(2019/09/10)
- htmlファイルの形式を「XHTML 1.0 Strict」から「html5」に変更しました。また、
Nu Html Checkerというhtmlの文法チェック結果を大幅に改善しました。
これに伴い、多少の不具合があるかもしれません。(2019/09/09)
- 「解析 | 機械学習(分類) | について」で基本的な考え方について、長ったらしくなりましたが記載しました。
まだざっくり版ですので、ミスはいくつか含んでいると思われます。(2019/09/07)
- 「サンプルデータ」51として、MLSeqを用いた
機械学習(分類/診断)を行う際の入力データを追加しました。(2019/09/06)
- 複数個所アップデートしてます。(2019/09/06)
- 「作図 | 生存曲線 | 基礎 | 2. pngファイルに保存」を追加しました。
3種類のデータを用いて、7つの例題を示しています。(2019/09/04)
- 「作図 | 生存曲線 | 基礎 | 1. まずはプロット」を更新しました。
3種類のデータを用いて、7つの例題を示しています。(2019/09/04)
- 「サンプルデータ」49-50として、生存曲線作成用のデータを追加しました。(2019/09/03)
- 「作図 | 生存曲線 | 基礎 | 1. まずはプロット」を追加しました。(2019/09/03)
- 「サンプルデータ」48として、生存曲線作成用のデータを追加しました。(2019/08/31)
- トーゴーの日シンポジウム2019が10月5日に開催されます。
申込締切は9月24日(火)24:00までです。(2019/08/27)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製なし | TCC(Sun_2013)」、
および「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC(Sun_2013)」で内部的に利用していたオプションを
"deseq2"から"deseq"に切り替えました。理由はDESeq2を使うとエラーが出るようになったからです(山本裕二郎 氏提供情報)。(2019/07/11)
- 3群間比較時に発現変動パターンまでうまく同定するための推奨パイプラインに関する論文が公開されました。
その手順は、「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+baySeq(Osabe_2019)」です。(2019/07/10)
- 「進化で読み解く バイオインフォマティクス入門(著:長田直樹)」が出版されています。
本書の何よりも素晴らしいところは、単著だという点だと思います(統一感って重要)。そしてチャラチャラしたところがなく、中身がしっかりしており、
そして幅広い内容が丁寧に解説されているという点が非常によいと思います。(2019/07/05)
- RNA-seqカウントデータ解析用RパッケージであるTCCのGUI版である、
TCC-GUIの論文 (Su et al., BMC Res Notes, 2019)がpublishされました。
利用法の英語版はAdditional file 2から取得可能です。
また、2群間比較用のリアルデータの解析例はAdditional file 3から取得可能です。
日本語版は、2019年3月15日の講義資料(の後半部分)に記載しています。(2019/03/14)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+EBSeq(Osabe_2019)」をアップデートしました。(2019/07/01)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+baySeq(Osabe_2019)」をアップデートしました。(2019/07/01)
- 「正規化 | 基礎 | TPM(Li_2010)」の原著論文を変更しました。(2019/06/25)
- 「解析 | クラスタリング | scRNA-seq | について」をアップデートしました。(2019/06/27)
- 「解析 | 前処理 | scRNA-seq | について」をアップデートしました。(2019/06/26)
- 「正規化 | 基礎 | TPM(Li_2010)」を追加しました。(2019/06/25)
- 「正規化 | 基礎 | RPKM(Mortazavi_2008)」の例題4にTPM (Transcripts Per Kilobase Million)との違いが分かり易いコードを追加しました。(2019/06/25)
- 「マッピング | について」をアップデートしました。(2019/06/04)
- 「イントロ | NGS | アノテーション情報取得 | について」をアップデートしました。(2019/06/04)
- 「前処理 | クオリティコントロール | について 」をアップデートしました。(2019/05/29)
- 「解析 | 発現量推定(トランスクリプトーム配列を利用)」をアップデートしました。(2019/05/24)
- 「マップ後 | カウント情報取得 | について」をアップデートしました。(2019/05/24)
- 「解析 | 発現変動 | について」をアップデートしました。(2019/05/24)
- 「正規化 | サンプル間 | について」をアップデートしました。(2019/05/23)
- 「解析 | 解析 | 融合遺伝子の同定」を追加しました。(2019/05/21)
- 「イントロ | NGS | 様々なプラットフォーム」をアップデートしました。(2019/05/21)
- 「アセンブル | トランスクリプトーム(転写物)用」をアップデートしました。(2019/05/21)
- 「解析 | 新規転写物同定(ゲノム配列を利用)」をアップデートしました。(2019/05/21)
- 「カウント情報取得 | シミュレーションデータ | scRNA-seq | 応用(異なる細胞群) | Splatter(Zappia_2017)」
を追加しました。(2019/04/11)
- 「カウント情報取得 | シミュレーションデータ | scRNA-seq | 基礎(異なる細胞群) | Splatter(Zappia_2017)」
を追加しました。(2019/04/11)
- 削除予定としていた「インストール | Rパッケージ | 必要最小限」を本当に削除しました。(2019/04/11)
- 削除予定としていた「インストール | Rパッケージ | ほぼ全て」を本当に削除しました。(2019/04/11)
- 「カウント情報取得 | シミュレーションデータ | scRNA-seq | 基礎(同一細胞群) | Splatter(Zappia_2017)」
を追加しました。(2019/04/11)
- 「解析 | 一般 | アラインメント | について」だった項目名を
「解析 | 一般 | アラインメント | ペアワイズ | について」と
「解析 | 一般 | アラインメント | マルチプル | について」に分離しました。(2019/04/05)
- 「カウント情報取得 | シミュレーションデータ | について」だった項目名を
「カウント情報取得 | シミュレーションデータ | RNA-seq | について」に変更しました。
また、「カウント情報取得 | シミュレーションデータ | scRNA-seq | について」も追加しました。(2019/04/05)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」を更新しました。(2019/04/06)
- 「解析 | 前処理 | scRNA-seq | について」を追加しました。(2019/04/04)
- 「解析 | クラスタリング | について」だった項目名を、
(bulk) RNA-seq用の「解析 | クラスタリング | RNA-seq | について」と、
scRNA-seq用の「解析 | クラスタリング | scRNA-seq | について」に変更しました。
それに伴い、中身や関連する項目名も変更しました。(2019/04/03)
- 「カウント情報取得 | シミュレーションデータ | について」を更新しました。(2019/04/03)
- 「カウント情報取得 | について」だった項目名を「カウント情報取得 | リアルデータ | について」
に変更しました。それに伴い、紹介するプログラムもリアルデータのもののみにしました。(2019/04/03)
- 2019年度もアグリバイオインフォマティクス教育研究プログラムを実施します。
例年東大以外の企業の方、研究員、大学院生が2割程度受講しております。受講ガイダンスは、2019年4月5日(Fri.)17:15より東大農学部2号館2階化学第一講義室で開催します。(2019/03/11)
- 細かいところの修正はここに明記していなくても随時行っています。(2019/03/11)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」を更新しました。(2019/03/08)
- 「生命科学データ解析を支える情報技術(監修:坊農秀雅)」が出版されています。
最先端のネタを含むかなり広範な内容を含んでいますので、一通り目次を眺めてみるとよいと思います。
Bioconda, Homebrew, Docker, GitHub, EC2, AWSなど聞いたことがある有用そうなものの全体像がわかるというメリットがあると思います。(2019/02/06)
- 「イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)」でエラーが出るようになっていたので修正しました。(2019/02/01)
- 2019年2月19日に「Rの講習会」を開催します。2019年2月1日15:00現在の申込状況:100名。(2019/02/01)
- 2018年
- 「生命科学データベース・ウェブツール(監修:坊農秀雅・小野浩雅)」が出版されています。
目次を一見すると既視感がありますが、実際に中身を見てみると”確かに手に取って読む価値がある”と判断できると思います。(2018/11/29)
- 「よくわかるバイオインフォマティクス入門(藤博幸 編)」が出版されています。
アラインメントの基本から深層学習までバイオインフォの幅広い内容が含まれています。(2018/11/21)
- TCCのオンラインGUI版(のベータ版)を公開しました。(2018/10/15)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ」を更新しました。(2018/11/13)
- 「インストール | Rパッケージ | 個別(2018年11月以降)」を変更しました。(2018/11/12)
- 永らく「削除予定」としていた項目(「Rのインストールと起動」、「個別パッケージのインストール」、「NOISeq(Tarazona_2011)」、「NBPSeq(Di_2011)」)を本当に削除しました。(2018/11/12)
- サブページに移行した項目を削除しました。(2018/11/12)
- 平成29年度のNGSハンズオン講習会でもお世話になった「先進ゲノム支援」による中級者向けの
情報解析講習会が2018年11月19-21日に開催されます。10/16締切です。(2018/09/20)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC正規化(Sun_2013)+EBSeq(Leng_2013)」
中の一部のコードが間違っていたので修正しました。(長部 高之 氏提供情報)(2018/08/12)
- 項目名「解析 | フィルタリング | について 」を「解析 | 前処理 | について 」に変更しました。
- 書ききれませんが、いろいろと追加や変更を行っています。(2018/08/08)
- 項目名「イントロ | 型変換 | ...」を「解析 | 前処理 | 型変換 | ...」に変更しました。(2018/08/06)
- 項目名「解析 | フィルタリング | について 」を「解析 | 前処理 | について 」に変更しました。
このあたりは、今後の情報増加に伴って、多少項目名を随時再編する予定です。(2018/08/06)
- 「前処理 | フィルタリング | について 」を追加しました。(2018/08/06)
- 「解析 | クラスタリング | サンプル間 | TCC(Sun_2013)」の例題10の入力ファイルリンク切れを修正しました。(2018/08/06)
- 「カウント情報取得 | リアルデータ | SRP001540 | recount(Collado-Torres_2017)」を更新しました。(2018/08/06)
- 「イントロ | 型変換 | について」を追加しました。(2018/08/02)
- 「イントロ | 型変換 | ExpressionSet --> SummarizedExperiment」を追加しました。(2018/08/02)
- 「イントロ | 型変換 | ExpressionSet --> RangedSummarizedExperiment」を追加しました。(2018/08/02)
- 「イントロ | 型変換 | RangedSummarizedExperiment --> ExpressionSet」を追加しました。(2018/08/02)
- 「カウント情報取得 | リアルデータ | ...」のところで、これまでRangedSummrizedExperimentオブジェクトをhogeとして取り扱ってきましたが、rseに変更しました。(2018/08/02)
- 「イントロ | 一般 | ExpressionSet | 1から作成 | NOISeq(Tarazona_2015)」を追加しました。(2018/08/02)
- 「イントロ | 一般 | ExpressionSet | 1から作成 | Biobase」を追加しました。(2018/08/01)
- 「カウント情報取得 | リアルデータ | SRP001540 | recount(Collado-Torres_2017)」を更新しました。例題4と5はその後の解析がやりやすいようにしています。(2018/07/31)
- 「カウント情報取得 | シミュレーションデータ | Biological rep. | 2群間 | 基礎 | LPEseq(Gim_2016)」のエラーが解消されました。(2018/07/31)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | NOISeq(Tarazona_2015)」を追加しました。(2018/07/29)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ(数GB?!)」を更新しました。(2018/07/29)
- 「カウント情報取得 | シミュレーションデータ | Biological rep. | 2群間 | 基礎 | LPEseq(Gim_2016)」を追加しました。今のところまだエラーが出ます。(2018/07/28)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | LPEseq(Gim_2016)」を追加しました。(2018/07/27)
- 「カウント情報取得 | シミュレーションデータ | ...」のあたりを追加しました。(2018/07/22)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | について」を追加しました。(2018/07/18)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | edgeR(Robinson_2010)」を更新しました。(2018/07/18)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | baySeq(Hardcastle_2010)」を更新しました。(2018/07/10)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | EBSeq(Leng_2013)」を更新しました。(2018/07/09)
- R version 3.5.1とversion 3.4.3でEBSeqパッケージを利用すべくlibrary(EBSeq)をしたら、
内部的に利用するblockmodelingというパッケージのCITATIONが原因でロードできないエラーに遭遇しました。
対処法はblockmodelingパッケージのフォルダ中にあるCITATIONというファイルの削除(私はこれでEBSeqをロードできるようになった)です。
blockmodelingフォルダのインストールされている場所がわからないヒトは、Windows2018.03.12版のスライド5などを参考にしてください。(2018/07/08)
- 「解析 | ChIP-seq | について」を更新しました。(2018/07/06)
- 「解析 | 機能解析 | GMTファイル取得 | について」を更新しました。(2018/07/06)
- 「カウント情報取得 | リアルデータ | SRP001540 | GSVAdata(Hänzelmann_2013)」を更新しました。(2018/07/03)
- 「解析 | 一般 | Sequence logos | ggseqlogo(Wagih_2017)」を(とりあえず項目のみ)追加しました。(2018/06/29)
- 「解析 | 一般 | Sequence logos | seqLogo」に項目名を変更しました。(2018/06/29)
- 「解析 | 一般 | Sequence logos | について」を追加しました。(2018/06/29)
- 「イントロ | NGS | アノテーション情報取得 | について」を更新しました。(2018/06/29)
- 「解析 | 機能解析 | GMTファイル取得 | について」を追加しました。(2018/06/27)
- 「解析 | 機能解析 | GMTファイル取得 | EGSEAdata(Alhamdoosh_2017)」を追加しました。(2018/06/27)
- 「解析 | 機能解析 | GMTファイル取得 | GeneSetDB(Araki_2012)」を追加しました。(2018/06/27)
- 「解析 | クラスタリング | について」を更新しました。(2018/06/27)
- 「解析 | 発現変動 | 時系列 | について」を更新しました。(2018/06/27)
- 「解析 | 分類 | について」を追加しました。(2018/06/27)
- 「解析 | 機能解析 | パスウェイ(Pathway)解析 | GSVA(Hänzelmann_2013)」を追加しました。(2018/06/26)
- 「解析 | 機能解析 | 遺伝子オントロジー(GO)解析 | GSVA(Hänzelmann_2013)」を追加しました。(2018/06/26)
- 「解析 | 機能解析 | 遺伝子セット解析 | GSVA(Hänzelmann_2013)」を更新しました。(2018/06/26)
- 「解析 | 機能解析 | 遺伝子オントロジー(GO)解析 | SeqGSEA(Wang_2014)」の記載事項を少し変更しました。
具体的には、MSigDBからのgmtファイル取得に関する別項目を新たに作成したので、この中に記載していた該当部分を大幅に削りました。(2018/06/25)
- 「解析 | 機能解析 | GMTファイル読込 | GSEABase(Morgan_2018)」を追加しました。(2018/06/25)
- 「解析 | 機能解析 | GMTファイル取得 | MSigDB(Subramanian_2005)」を追加しました。(2018/06/25)
- 「解析 | 機能解析 | について」を追加しました。GSEA周辺の歴史や考え方、
そしてどのようにして必要な情報を取得し解析するかについて、全貌をざっくりと書いてあります。(2018/06/25)
- 「カウント情報取得 | リアルデータ | SRP001540 | GSVAdata(Hänzelmann_2013)」を追加しました。(2018/06/22)
- 「カウント情報取得 | リアルデータ | SRP001540 | recount(Collado-Torres_2017)」を追加しました。(2018/06/22)
- 「インストール | Rパッケージ | 必要最小限プラスアルファ(数GB?!)」を更新しました。(2018/06/22)
- 「解析 | 機能解析 | 遺伝子オントロジー(GO)解析 | について」を更新しました。(2018/06/22)
- 「解析 | 機能解析 | パスウェイ(Pathway)解析 | について」を更新しました。(2018/06/22)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | Blekhmanデータ | TCC(Sun_2013)」を追加しました。(2018/06/18)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC(Sun_2013)」の項目を追加しました。(2018/06/17)
- 以下の2つの項目は、Rパッケージrecountを用いて
ウェブサイトrecount2にアクセスしてカウント情報を含む
RangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードして、各種カウントデータを抽出したりするやり方を示しています。
メタデータが公共DBに依存しており一筋縄ではいきませんので、ERP000546とSRP001558のやり方を見比べて一通りの例題をこなして経験を積んでおくことを強く推奨します。
私はまだrecount2の原著論文を読んではおりませんが、提供されている生物種はおそらくヒトのみです。
それでもなお、統一的な手順で得られたカウントデータを提供してくれてますので、様々なデータセットを直接比較できるというadvantageは非常に大きいと思います。(2018/06/10)
- 「カウント情報取得 | リアルデータ | ERP000546 | recount(Collado-Torres_2017)」を追加しました。(2018/06/10)
- 「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」を追加しました。(2018/06/10)
- 「カウント情報取得 | について」を追加しました。(2018/06/10)
- 「マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | HTSeq(Anders_2015)」の中身を変更しました。(2018/06/06)
- 「マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | HTSeq(Anders_2015)」を追加しました。(2018/05/30)
- 「イントロ | ファイル形式の変換 | GFF3 --> GTF」を追加しました。(2018/05/30)
- 「H29年度NGSハンズオン講習会」の報告書が公開されました。(2018/05/17)
- Silhouetteスコアの新たな使い道提唱論文(Zhao et al., Biol. Proc. Online, 2018)
の使い方を「解析 | 一般 | Silhouette scores(シルエットスコア)」に示しました。(2018/03/01)
- Silhouetteスコアの新たな使い道提唱論文(Zhao et al., Biol. Proc. Online, 2018)がpublishされました。(2018/03/01)
- アグリバイオインフォマティクス教育研究プログラムでは、平成30年度もバイオインフォ関連講義を行います。
例年東大以外の企業の方、研究員、大学院生が2-3割程度受講しております。受講ガイダンスは、平成30年4月4日17:15より東大農学部2号館2階化学第一講義室で開催します。(2018/03/08)
- 「作図 | M-A plot | 基礎 | 2. 発現変動遺伝子を色分けする」の例題5で示しているように、
TCCの推奨手順(内部的にDESeq2を利用)で複製なしデータの発現変動解析を行ったときに、
明らかにおかしな結果になる場合があることが判明しましたのでお知らせします(南茂隆生 氏提供情報)。
私がこれまで動作確認用で用いてきた複製なしデータ(data_hypodata_1vs1.txt)ではうまくいっていたので、
今まで全くこの問題に気づきませんでした。私どもを信頼してTCCを利用して頂いていた皆さま、大変申し訳ありませんでしたm(_ _)m
もう少し詳細な内容については例題5のところにも記載しております。
もしM-A plotで眺めて「明らかに変」な結果に遭遇した方が他にいらっしゃいましたら、ご連絡いただければ幸いです。
尚、当面の対策としては、とりあえずは現状の推奨手順でやっていただいて、M-A plotが変じゃなければそのままでよいと思います。
もし変な結果が得られれば、内部的にDESeq2ではなくDESeqを用いるTCCの利用で満足のいく結果が得られるものと期待されます。
大変貴重な情報をお寄せいただいた南茂隆生 氏、およびDESeq2開発者とのやりとりや現象の把握に尽力してくださった孫建強 氏に感謝申し上げますm(_ _)m。
尚、私の動作環境はWindows, R ver. 3.3.3, TCC ver. 1.14.0, DESeq2 ver. 1.14.1です。(2018/01/12)
- 「作図 | M-A plot...」のあたりが2012年頃の古い記述のままになっていたので更新しました。(2018/01/11)
- 「サンプルデータ」を更新しました。例題45と46です(孫建強氏 提供情報)。
ついでに例題44のrecount2の論文
(Collado-Torres et al., Nat Biotechnol., 2017)情報を更新しました。(2018/01/11)
- 2017年
- Dr. Bonoの生命科学データ解析
というバイオインフォマティクス初学者向けの本が出版されています。この教科書を読めばバイオインフォの基礎知識や基本的な考え方などを体系的に学べます。
NGSハンズオン講習会で専門用語などについていけなかったヒトは、この教科書で基礎知識を補っておくとよいと思います。(2017/10/06)
- 「参考資料 | 講習会、講義、講演資料」を更新しました。(2017/09/07)
- R (ver. 3.4.1; 2017年5月以降ごろから使えるやつ)のTCCパッケージ利用時
に、Macユーザの方がエラーが出るようです。対策は、「R ver. 3.3.3などちょっと古いバージョンのものを使う」です。
原因はTCC内部で利用しているsamrパッケージにバグが含まれており(2017年7月31日現在)、
これをインストールできないからです。お気を付けください(マックユーザ2名からの提供情報)。(2017/07/31)
- 「イントロ | 一般 | 配列取得 | プロモーター配列 | GenomicFeatures(Lawrence_2013)」の例題12で、
コンティグ(配列)数が複数で「FASTAファイルには存在するがGFFファイル中には存在しない配列があった場合」に不都合が生じる問題を回避できるコードに書き換えました(野間口達洋氏 提供情報)。(2017/06/23)
- 解析 | 菌叢解析 | についてをアップデートしました。(2017/06/04)
- Galaxyのウェブサイトのリンク先をhttp://usegalaxy.org/からhttps://galaxyproject.org/に変更しました。(2017/03/17)
- 私の所属するアグリバイオインフォマティクス教育研究プログラムでは、平成29年度もバイオインフォ関連講義を行います。
例年東大以外の企業の方、研究員、大学院生が2-3割程度受講しております。受講ガイダンスは4月5日17:15- 於東大農です。
例年アグリバイオ所有ノートPCは台数が絶対的に足りないので、特に外部の受講希望者はできるだけ基本的に3時間以上バッテリーがもつノートPCを用意して臨んで下さい。(2017/02/20)
- 2016年
- 「解析 | 一般 | アラインメント | ...」周辺の項目名を整理しました。(2016/12/29)
- 2016年10月5-6日に東京大学弥生講堂一条ホールにてトーゴーの日シンポジウム2016
が開催されます。主催はNBDC!。NGSハンズオン講習会では裏方に徹して事前準備から後片付けまで大変お世話になりましたm(_ _)m。
講習会受講者アンケートで書かれていた希望や要望の一部はポスター発表者への質問で解決するかもしれません。
興味ある方は是非ご参加ください。(2016/09/30)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製なし | DESeq2(Love_2014)」を作成しました。(2016/06/01)
- 「解析 | クラスタリング | 遺伝子間(応用) | TCC正規化(Sun_2013)+MBCluster.Seq(Si_2014)」
のリンク先が切れていたのを修正しました。コードも若干変更しました。(2016/05/30)
- これまでずっと放置していた「FDR < 閾値」という変な表現を、やっと「FDR = 閾値 (q-value < 閾値)」に修正しはじめました。
該当箇所は多数あります(爆)。(2016/05/23)
- 「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | DESeq2(Love_2014)」を作成しました。
DESeq2のq-valueと、DESeq2のp-valueからp.adjust関数を用いて得られたq-value (adjusted p-value)に大きく違いが出るデータに初めて遭遇し、私も驚いています。(2016/05/22)
- 解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC(Sun_2013)で、内部的に用いる方法をDESeqからDESeq2に変更しました。(2016/05/21)
- R ver. 3.3.0、およびBioconductor ver. 3.3がリリースされています。NGSハンズオン講習会の講義資料はこのバージョンで作成、動作確認予定です。(2016/05/10)
- ウェブページが大きくなりすぎて重いのは承知しておりますw。2016年8月のNGSハンズオン講習会後に、ページを2分割予定ですm(_ _)m(2016/04/27)
- 「イントロ | 一般 | 任意のキーワードを含む行を抽出(基礎)」で、
例題4以降の多くのものについて、不具合修正やコメントを追加しました(アグリバイオ受講生提供情報)。(2016/04/20)
- QuasRでBowtieのマッピングを行う場合に、
(内部的にはbowtie1が動いているため)リード長が1本でも1,024 bpを超えたものがあればコケマス(1024 bpはセーフで1025 bpはアウト)のでご注意ください(高橋 広夫 氏提供情報)。(2016/04/06)
- RNA-Seq実験ハンドブック(鈴木穣 編)が刊行されます。(2016/03/22)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC正規化(Sun_2013)+baySeq(Hardcastle_2010)」
が一通り動くようになりました。TCC正規化を含めることでAUC値(感度・特異度)が上がっているところまでは確認済みです。(2016/03/13)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC正規化(Sun_2013)+EBSeq(Leng_2013)」
が一通り動くようになりました。TCC正規化を含めることでAUC値(感度・特異度)が上がっているところまでは確認済みです。(2016/03/13)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | baySeq(Hardcastle_2010)」
が一通り動くようになりました。(2016/03/07)
- マッピングやカウント情報取得の周辺項目をアップデートしています。(2016/02/12)
- 使えなくなった関数名makeTranscriptDbFromGFFをmakeTxDbFromGFFに全て変更しました。
これに関連するTranscriptDbという記述をTxDbに変更しました。(2016/02/09)
- Erratum。2014.06.25のアグリバイオ大学院講義資料中で
「アセンブル結果で平均コンティグ長やN50を大きくするには、kの値を小さめにすればいい」
と全く逆のことを書いていることに、今頃気づきました。正しくは大きめにすればいいです。
(見てるヒトはほとんどいないと思いますがw)念のため修正版に差し替えてあります。大変失礼しましたm(_ _)m(2016/02/01)
- 2015年
- 解析 | 一般 | アラインメント | についてを追加しました。(2015/12/16)
- xlsx形式ファイルを読み込むやり方を「イントロ | 一般 | 読み込み | xlsx形式 | openxlsx」に示しました。(2015/11/15)
- 2015年4-10月初旬頃Bioconductor提供パッケージ群をインストールしていた方は、
おそらくR本体のバージョンがR ver. 3.2.0(2015-04-16リリース)またはver. 3.2.1(2015-06-18リリース)だろうと思います。
この方々が、私の推奨手順通りに2015年11月にR ver. 3.2.2(2015-08-14リリース)でパッケージのアップデートをする際に
遭遇するかもしれないエラーとその対処法をインストール | についてに示しました。(2015/11/12)
- 多群間比較用の推奨ガイドライン提唱論文(Tang et al., BMC Bioinformatics, 2015)がpublishされました。
論文概要については門田のページでも紹介しています。
講習会でよく述べている「サンプル間クラスタリング結果からDEG検出結果のおおよその見積もりが可能である」
という主張の根拠となる原著論文がこれになります。推奨ガイドライン周辺の関連項目もアップデートしました。(2015/11/05)
- sample1.fastaのような配列が1つしかない場合に、
rowSums(x)の計算時にエラーが出ることがわかったので、該当箇所をapply(as.matrix(x), 1, sum)のような感じに変更しました。(2015/09/12)
- 解析 | 発現変動 | 時系列 | maSigPro(Nueda_2014)が一通りできるようになりました。
まだ完全には結果を解釈しきれていませんが、「fit$SELEC」で見えているものがDEGの発現パターンであり、「out$summary」がDEGのIDリストです。(2015/08/16)
- イントロ | 一般 | 配列取得 | プロモーター配列 | GenomicFeatures(Lawrence_2013)の例題5で、
取得したい領域がsense, antisense両方ともゲノム配列の範囲内に収まるものだけを抽出して出力するよりよいコードに書き換えました(甲斐政親氏 提供情報)。(2015/08/14)
- イントロ | 一般 | 配列取得 | プロモーター配列 | GenomicFeatures(Lawrence_2013)の例題4で、
取得したい領域がsense, antisense両方ともゲノム配列の範囲内に収まるものだけを抽出して出力するよりよいコードに書き換えました(甲斐政親氏 提供情報)。
他の部分についても順次反映させていきます。(2015/08/04)
- 前処理 | クオリティチェック | Overrepresented sequences | ShortRead(Morgan_2009)の「ファイルに保存」の部分で
「tmp <- cbind(names(out), out)」と書くのは冗長であるとの指摘を受けたのでその周辺を修正しました(野間口達洋氏 提供情報)。(2015/07/29)
- 前処理 | フィルタリング | paired-end | 配列長とN数 | QuasR(Gaidatzis_2015)を作成し、
リード数が同じpaired-endデータを入力として許容する最短配列長やN数でフィルタリングするやり方を示しました(Thanks to Dr. Stadler)。(2015/06/26)
- 前処理 | トリミング | アダプター配列除去(応用) | QuasR(Gaidatzis_2015)で
QuasRの枠組みでpaired-endデータを処理する手順を示しました(Thanks to Dr. Stadler)。(2015/06/26)
- 「前処理 | トリミング」と「前処理 | フィルタリング」の順番を入れ替えました。(2015/06/26)
- 前処理 | フィルタリング | paired-end | 共通リード抽出 | ShortRead(Morgan_2009)で
リード数の異なるpaired-endデータへの対応が完了しました。(2015/06/26)
- 前処理 | トリミング | アダプター配列除去(基礎) | QuasR(Gaidatzis_2015)などで
param_nrecが適切に反映されるように修正しました(中村浩正 氏提供情報)。(2015/06/24)
- イントロ | NGS | 読み込み | FASTQ形式 | 基礎の例題8でNAへの対処法を追加しました(茂木朋貴氏、野間口達洋氏、他多くの受講生提供情報)。(2015/06/23)
- 「前処理 | トリミング | アダプター配列除去」周辺の項目を更新しました。(2015/06/23)
- イントロ | NGS | 読み込み | FASTQ形式 | 応用でgzip圧縮FASTQファイルから
メモリを消費せず(一旦全部読み込むことをせずに)にサブセットを取り出す方法を示しました(野間口達洋 氏提供情報)。(2015/06/18)
- 前処理 | クオリティチェック | QuasR(Gaidatzis_2015)の項目を追加しました。(2015/06/15)
- 「イントロ | NGS | 読み込み | BSgenome | 基本情報を取得」
でマウスやヒトゲノムを解析する際に生じていた「整数オーバーフロー問題」を回避するコードに変更しました(野間口達洋 氏提供情報)。(2015/05/27)
- Rパッケージのインストールで、RobLoxBioC
パッケージが2015年現在のR本体の最新リリースに対応していないとのことでリストから外しました。(2015/05/25)
- Rパッケージのインストール周辺で、limma
(Ritchie et al., Nucleic Acids Res., 2015)をBioconductorから
取得しないといけないにもかかわらずCRANから取得しようとしてこけていたのに気づきました。修正済みですm(_ _)m(2015/05/25)
- 「アセンブル | ゲノム用」の情報を更新しました。(2015/05/15)
- パッケージのインストールのところで、SAFEではなくsafeパッケージであるというご指摘をいただきましたので該当箇所を修正しました(野間口達洋 氏提供情報)。(2015/04/24)
- 「アセンブル | トランスクリプトーム(転写物)用」の情報を更新しました。(2015/04/22)
- 私の所属するアグリバイオインフォマティクス教育研究プログラムでは、平成27年度もバイオインフォ関連講義を行います。
例年東大以外の企業の方、研究員、学生が2-3割程度受講しております。受講ガイダンスは4月6日17:15- 於東大農です。(2015/03/31)
- R本体およびパッケージのインストール手順のところを更新しました。詳細はインストール | についてをごらんください。(2015/04/02)
- MBCluster.Seqパッケージを用いた遺伝子間クラスタリングのやり方を一通り示しました。(2015/03/14)
- 翻訳配列取得において、seqinrパッケージを用いてtranslate関数を実行するほうが
翻訳できないコドンはアミノ酸X(不明なアミノ酸)に変換してくれたり、ambiguous=Tオプションを利用することで翻訳できるものは可能な限り翻訳してくれる(高橋 広夫 氏提供情報)
ということで、周辺情報を追加しました。lapply関数を用いるやり方(高橋 広夫 氏提供情報)とsapply関数を用いるやり方(甲斐 政親 氏提供情報)を示しています。(2015/03/09)
- QuasRパッケージを用いてマッピングをする際に、getwd()で見られる
パス名の中に日本語が含まれているとエラーが出る(高橋 広夫 氏提供情報)とのお知らせをいただきました。ご注意ください。(2015/03/08)
- seqinrパッケージの原著論文(Charif et al., Bioinformatics, 2005)を2007年のものから変更しました。(2015/03/08)
- TxDb周辺情報で、GFFファイルの読み込み時にChrCが環状ゲノムと指定するやり方(高橋 広夫 氏提供情報)を追加しました。(2015/03/04)
- 「作図 | クラスタリング」周辺の情報を追加しました。(2015/02/15)
- 「作図 | ROC曲線」周辺で、発現変動ランキング結果のROC曲線やAUC値の感覚を理解するための例題を充実させています。(2015/02/08)
- 「解析 | 発現変動 | 3群間 | 対応なし | 複製あり」周辺の情報を追加しました。(2015/02/04)
- 「解析 | シミュレーションカウントデータ」周辺で、発現変動解析時に動作確認用として用いるシミュレーションカウントデータを自在に作成するための項目を充実させつつあります。(2015/01/25)
- QuasRパッケージ(Gaidatzis et al., Bioinformatics, 2015)
中のextractTranscriptsFromGenome関数実行部分でエラーが出るようです(QuasR ver. 1.6.2あたり; Bioconductor 3.0)。対策として当該関数をとりあえずextractTranscriptSeqsに変更しました。(2015/01/21)
- 2014年
- 門田幸二 著シリーズ Useful R 第7巻 トランスクリプトーム解析、およびこのウェブページ中で頻用させていただいているQuasR
パッケージの原著論文(Gaidatzis et al., Bioinformatics, 2015)が公開されたので関連個所をアップデートしました。(2014/12/03)
- 「解析 | ChIP-seq | について」の情報を少しアップデートして、
実験医学2014年12月号にも掲載されているSraTailor (Oki et al., Genes Cells., 2014)の情報などを追加しました。(2014/11/25)
- ウェブサーバ引っ越し作業のため、11/25前後でダウン予定(最長で11/21-26)です。(2014/11/06)
- NGS解析に限らず、Rに限らず、多くの研究者が持っている一通りのデータ解析に関する解説付きのコマンド集を充実させていければと思っています。
とりあえずこのサイトでは(○○氏 提供情報)とさせていただきますので、情報をお寄せいただければ幸いです。
長期的にはこのサイトでなくてもいいので日本全体のノウハウ集や教材を統合DB的に集約するような枠組みになればと思っています。(2014/09/27)
- 2014年10月04日にHPCIワークショップ「医療とビッグデータ解析」(9:00-9:20)に引き続いて
中級者向けバイオインフォマティクス入門講習会@仙台国際センター(10:50-12:20)で話します。
スライド中のhogeフォルダの圧縮ファイルはhoge.zip(20140929, 22:27版)です。
20140819版から、htmlのスタイルファイル情報を追加して見栄えをよくしただけです(2014/09/29)
- 入出力のファイル名について、FASTA形式ファイルの拡張子は.fasta、FASTQ形式ファイルの拡張子は.fastqに変更する作業がほぼ完了しております。(2014/07/17)
- 配列長とカウント数の関係のところで、
boxplotでの描画の際にparam個で分割(20分割など)するテクニックとして「floor(nrow(data)/param)+1」としていましたが、
これだと割り切れる場合でも+1してしまうことが判明したため「ceiling(nrow(data)/param)」に修正しました(佐伯亘平氏提供情報)。(2014/07/03)
- 2014年07月22日にイルミナウェビナーで話します。興味ある方はどうぞ。(2014/06/30)
- writeFastq関数のデフォルトがgzip圧縮(孫建強氏提供情報)であることが分かったので関連項目を修正しました。
これに関連して、FASTA形式ファイルの拡張子は.fasta、FASTQ形式ファイルの拡張子は.fastqに順次変更していきます。(2014/06/15)
- 2014年06月12日にNAIST植物グローバル教育プロジェクト・平成26年度ワークショップ「ImageJ+Rハンズオン実習2014」
が開催されます。特に門田の部分を受講したい方は2014年4月22日に作成したより詳細なインストール手順(Windows版)を参考にしてインストールしておいてください。
Mac版のインストール手順(by 孫建強氏)もあります。
Macのヒトの注意点は、「Mac OS X のバージョンに関わらず R-3.1.0-snowleopard.pkg をインストールしたほうがよい」です。
また、実習用データ(hoge.zip; 約40MB)もダウンロードしておいてください。(2014/05/14)
- 機能解析の遺伝子オントロジー(GO)解析とパスウェイ(Pathway)解析周辺を更新し、SeqGSEAパッケージを用いた解析のみですが一通りできるようにしました。(2014/03/30)
- 私の所属するアグリバイオインフォマティクス教育研究プログラムでは、平成26年度も(東大生に限らず)バイオインフォ関連講義を行います。
4/9に私の第一回目の講義がありましたが、過去最高の123名の出席がありました。例年東大以外の企業の方、研究員、学生が二割程度は受講しております。
このウェブページと直接関連する講義は「ゲノム情報解析基礎」と
「農学生命情報科学特論I」ですが、背景理論の説明などは「機能ゲノム学」でも行います。
興味ある科目のみの受講も可能ですので、お気軽にどうぞ。(2014/04/10)
- 一連の解析パイプライン(RNA-seqデータ取得 -> マッピング -> カウントデータやRPKMデータ取得 -> サンプル間クラスタリングや発現変動解析およびM-A plot描画まで)のクラスタリング部分をアップデートしました。項目名の一番下のほうです。(2014/02/26)
- 2014年3月17-19日に九州大学にて、ワークショップ(よく分かる次世代シークエンサー解析 ~最先端トランスクリプトーム解析~)が開催されます。
私は3日目(3/19, 13:00-16:30)を担当します。興味ある方はどうぞ。締切は確か2/21です。(2014/02/17)
- 項目名の整理を行っています。3C (Hi-C)やBS-seq周辺についても少し言及してあります。(2014/02/08)
- 2013年
- 発現変動解析用RパッケージTCC (ver. 1.2.0; Sun et al., BMC Bioinformatics, 2013)がBioconductorよりリリースされました。
最新版を利用したい方は、R (ver. 3.0.2)をインストールしたのち、Bioconductor (ver. 2.13)をインストールしてください。(2013/10/17)
- どのブラウザからでもエラーなく見られる(W3C validation)ように((Rで)マイクロアレイデータ解析も含めて)リニューアルしました。(2013/07/30)
- 2013年7月29日まで公開していた以前の「(Rで)塩基配列解析」のウェブページや関連ファイルはRdeennki.zipからダウンロード可能です(110MB程度)。(2013/07/30)
- 平成26年3月7日に東京お台場にて、HPCIチュートリアルの一部としてRでゲノム・トランスクリプトーム解析を行います。情報はかなりアップデート予定ですが、既にキャンセル待ちなようですみませんm(_ _)m(2013/11/25)
- 2013年6月6日に開催されたNAIST植物グローバル教育プロジェクト・平成25年度ワークショップ
のときに利用した、R(ver. 3.0.1)とTCC(ver. 1.1.99)などのインストール方法はこちら(Windows用のみ;hoge.zipはおまけ)です。
- 平成25年6月27日、7月3, 4日にこのウェブページ関連の実習を含む講義(農学生命情報科学特論I)を行います。
東大生以外の外部の方も受講可能です。詳しくは事務局までお問い合わせください。(2013/06/08)
- 廃止予定の関数名(read.DNAStringSet -> readDNAStringSetなど)や「前処理 | 正規化...」周辺の項目名の変更をしました。(2013/01/16)
- 2012年
- htmlのタグに問題があるらしくfirefoxでエラーという指摘をTbT論文共著者の西山さんから受けましたのでその周辺を修正しました。(2012/11/15)
- R2.15.2がリリースされていたのでこれに変更しました。(2012/11/15)
- 若干項目名を(あまりにも場違いだったものを)変更しました、直接リンクを張ってたかた、すみませんm(_ _)m。(2012/07/12)
インストール | について
以下は、「インストール | R本体とRStudio | 最新版」と「インストール | Rパッケージ | 必要最小限プラスアルファ」の推奨インストール手順をまとめたものです。
私の環境は、Windows PCは(Windows 10; 64 bit)、Macintosh PCはMacBook Pro (MacOS Monterey Ver.12.3.1; 64 bit)です。
インストール | R本体とRStudio | 最新版 | Win用
最新版(リリース版のこと)は、下記手順を実行します。インストールが無事完了したら、
デスクトップに「R x64 4.X.Y」アイコンが作成されます(XやY中の数値はバージョンによって異なります)。
2022年05月01日現在の最新版は、R-4.2.0-win.exeです。
- Rのインストーラを「実行」
- 基本的には「次へ」などを押しながらインストールを完了させる
- 「コントロールパネル」−「デスクトップのカスタマイズ」−「フォルダオプション」−「表示(タブ)」−「詳細設定」のところで、
「登録されている拡張子は表示しない」のチェックを外してください。
- RStudioのダウンロードサイトをクリックし、
「RStudio-2022.02.1-461.exe」と酷似したファイル名のものをクリック。
インストール | R本体とRStudio | 最新版 | Mac用
最新版(リリース版のこと)は、下記手順を実行します。インストールが無事完了したら、
Finderを起動して、左のメニューの「アプリケーション」をクリックすると、Rのアイコンが作成されていることが確認できます。
2022年05月16日現在の最新版は、R-4.2.0.pkgです。
- http://cran.r-project.org/bin/macosx/の「R-4.X.Y.pkg」をクリック。
(XやY中の数値はバージョンによって異なります)
- ダウンロードしたファイルをダブルクリックして、基本的には「次へ」などを押しながらインストールを完了させる
- 「Finder」-「環境設定」-「詳細」タブのところで「すべてのファイル名拡張子を表示」にチェックを入れる。
- RStudioのダウンロードサイトをクリックし、
「RStudio-2022.02.1-461.dmg」と酷似したファイル名のものをクリック。
- XQuartzをインストール(2022/05/16追加)
インストール | R本体 | 過去版 | Win用
昔のバージョンをインストールしたい局面もごく稀にあると思います。
その場合は、ここをクリックして、
任意のバージョンのものをインストールしてください。例えば、2014年10月リリースのver. 3.1.2をインストールしたい場合は、
3.1.2をクリックして、
「Download R 3.1.2 for Windows」
をクリックすれば、後は最新版と同じです。
インストール | Rパッケージ | について
アグリバイオで所有するノートPCは、基本的に
「インストール | Rパッケージ | 必要最小限プラスアルファ」
を利用してパッケージ群を一度にインストールしています。しかし、コロナ禍や講義で用いるパッケージの変遷などを経て、多少の不具合を許容しつつシンプルにインストールできる方針に変更しました(2022年3月30日)。
「インストール | Rパッケージ | 個別(2018年11月以降)」のところは、
インストールされていない(or されなかった)パッケージを個別にインストールする際に利用してください。
インストール | Rパッケージ | 必要最小限プラスアルファ
アグリバイオで所有するノートPCは、Rパッケージの2大リポジトリであるCRANと
Bioconductor(およびGithub)から提供されている以下のパッケージ群をインストールしています。
30分程度でインストールが完了します(自宅の光の無線LAN環境)。
1. RStudioを起動
2. パッケージ群のインストール
以下を「R コンソール画面」上でコピー&ペースト。
どこからダウンロードするか?と聞かれるので、その場合は自分から近いサイトを指定。
「no」の行に対するエラーは気にしなくて大丈夫です(Mac対応です)。
if (!requireNamespace("BiocManager", quietly=T))
install.packages("BiocManager")
BiocManager::install("ape", update=F)
BiocManager::install("bio3d", update=F)
BiocManager::install("blockmodeling", update=F)
BiocManager::install("bit", update=F)
BiocManager::install("cclust", update=F)
BiocManager::install("class", update=F)
BiocManager::install("cluster", update=F)
BiocManager::install("clValid", update=F)
BiocManager::install("corrplot", update=F)
BiocManager::install("data.table", update=F)
BiocManager::install("devtools", update=F)
BiocManager::install("dplyr", update=F)
BiocManager::install("DT", update=F)
BiocManager::install("e1071", update=F)
BiocManager::install("fansi", update=F)
BiocManager::install("ff", update=F)
BiocManager::install("fields", update=F)
BiocManager::install("FinePop", update=F)
BiocManager::install("FinePop2", update=F)
BiocManager::install("FIT", update=F)
BiocManager::install("fitdistrplus", update=F)
BiocManager::install("GeneCycle", update=F)
BiocManager::install("GGally", update=F)
BiocManager::install("glmnet", update=F)
BiocManager::install("gptk", update=F)
BiocManager::install("GSA", update=F)
BiocManager::install("heatmaply", update=F)
BiocManager::install("kernlab", update=F)
BiocManager::install("KernSmooth", update=F)
BiocManager::install("knitr", update=F)
BiocManager::install("mapdata", update=F)
BiocManager::install("maps", update=F)
BiocManager::install("MASS", update=F)
BiocManager::install("microseq", update=F)
BiocManager::install("mixOmics", update=F)
BiocManager::install("MVA", update=F)
BiocManager::install("openxlsx", update=F)
BiocManager::install("Peptides", update=F)
BiocManager::install("phateR", update=F)
BiocManager::install("plotly", update=F)
BiocManager::install("PoissonSeq", update=F)
BiocManager::install("pvclust", update=F)
BiocManager::install("qqman", update=F)
BiocManager::install("R6", update=F)
BiocManager::install("randomForest", update=F)
BiocManager::install("RColorBrewer", update=F)
BiocManager::install("rclipboard", update=F)
BiocManager::install("RCurl", update=F)
BiocManager::install("rentrez", update=F)
BiocManager::install("rgl", update=F)
BiocManager::install("rmarkdown", update=F)
BiocManager::install("rrBLUP", update=F)
BiocManager::install("Rtsne", update=F)
BiocManager::install("samr", update=F)
BiocManager::install("scatterplot3d", update=F)
BiocManager::install("seqinr", update=F)
BiocManager::install("shiny", update=F)
BiocManager::install("shinyBS", update=F)
BiocManager::install("shinycssloaders", update=F)
BiocManager::install("shinydashboard", update=F)
BiocManager::install("shinyWidgets", update=F)
BiocManager::install("som", update=F)
BiocManager::install("st", update=F)
BiocManager::install("survminer", update=F)
BiocManager::install("tidyverse", update=F)
BiocManager::install("umap", update=F)
BiocManager::install("varSelRF", update=F)
BiocManager::install("xfun", update=F)
BiocManager::install("zeallot", update=F)
BiocManager::install("zoo", update=F)
BiocManager::install("affy", update=F)
BiocManager::install("agilp", update=F)
BiocManager::install("annotate", update=F)
BiocManager::install("ArrayExpress", update=F)
BiocManager::install("baySeq", update=F)
BiocManager::install("beadarray", update=F)
BiocManager::install("BeadDataPackR", update=F)
BiocManager::install("betr", update=F)
BiocManager::install("BHC", update=F)
BiocManager::install("biomaRt", update=F)
BiocManager::install("Biostrings", update=F)
BiocManager::install("BSgenome", update=F)
BiocManager::install("bsseq", update=F)
BiocManager::install("Category", update=F)
BiocManager::install("ChIPpeakAnno", update=F)
BiocManager::install("chipseq", update=F)
BiocManager::install("ChIPseqR", update=F)
BiocManager::install("ChIPsim", update=F)
BiocManager::install("clusterStab", update=F)
BiocManager::install("cosmo", update=F)
BiocManager::install("CSAR", update=F)
BiocManager::install("dada2", update=F)
BiocManager::install("DECIPHER", update=F)
BiocManager::install("DEGseq", update=F)
BiocManager::install("DESeq", update=F)
BiocManager::install("DESeq2", update=F)
BiocManager::install("DiffBind", update=F)
BiocManager::install("doMC", update=F)
BiocManager::install("EDASeq", update=F)
BiocManager::install("edgeR", update=F)
BiocManager::install("EGSEA", update=F)
BiocManager::install("EGSEAdata", update=F)
BiocManager::install("gage", update=F)
BiocManager::install("genefilter", update=F)
BiocManager::install("GenomicAlignments", update=F)
BiocManager::install("GenomicFeatures", update=F)
BiocManager::install("GEOquery", update=F)
BiocManager::install("ggplot2", update=F)
BiocManager::install("girafe", update=F)
BiocManager::install("GLAD", update=F)
BiocManager::install("golubEsets", update=F)
BiocManager::install("GSAR", update=F)
BiocManager::install("GSEABase", update=F)
BiocManager::install("GSVA", update=F)
BiocManager::install("GSVAdata", update=F)
BiocManager::install("Heatplus", update=F)
BiocManager::install("illuminaMousev2.db", update=F)
BiocManager::install("impute", update=F)
BiocManager::install("limma", update=F)
BiocManager::install("lumi", update=F)
BiocManager::install("marray", update=F)
BiocManager::install("maSigPro", update=F)
BiocManager::install("MBCluster.Seq", update=F)
BiocManager::install("MLSeq", update=F)
BiocManager::install("msa", update=F)
BiocManager::install("Mulcom", update=F)
BiocManager::install("multtest", update=F)
BiocManager::install("NOISeq", update=F)
BiocManager::install("htSeqTools", update=F)
BiocManager::install("NBPSeq", update=F)
BiocManager::install("OCplus", update=F)
BiocManager::install("org.Hs.eg.db", update=F)
BiocManager::install("parathyroidSE", update=F)
BiocManager::install("pathview", update=F)
BiocManager::install("pcaMethods", update=F)
BiocManager::install("pcot2", update=F)
BiocManager::install("pd.rat230.2", update=F)
BiocManager::install("PGSEA", update=F)
BiocManager::install("phyloseq", update=F)
BiocManager::install("PICS", update=F)
BiocManager::install("plier", update=F)
BiocManager::install("puma", update=F)
BiocManager::install("qrqc", update=F)
BiocManager::install("QuasR", update=F)
BiocManager::install("r3Cseq", update=F)
BiocManager::install("RankProd", update=F)
BiocManager::install("recount", update=F)
BiocManager::install("REDseq", update=F)
BiocManager::install("rMAT", update=F)
BiocManager::install("Rsamtools", update=F)
BiocManager::install("rtracklayer", update=F)
BiocManager::install("safe", update=F)
BiocManager::install("SAGx", update=F)
BiocManager::install("segmentSeq", update=F)
BiocManager::install("seqLogo", update=F)
BiocManager::install("ShortRead", update=F)
BiocManager::install("sigPathway", update=F)
BiocManager::install("SpeCond", update=F)
BiocManager::install("SPIA", update=F)
BiocManager::install("splatter", update=F)
BiocManager::install("SplicingGraphs", update=F)
BiocManager::install("SRAdb", update=F)
BiocManager::install("tweeDEseqCountData", update=F)
BiocManager::install("TCC", update=F)
BiocManager::install("topGO", update=F)
BiocManager::install("TxDb.Hsapiens.UCSC.hg38.knownGene", update=F)
BiocManager::install("vsn", update=F)
#devtools::install_github("andrewsali/plotlyBars", upgrade=F)
#devtools::install_github("aroneklund/beeswarm", upgrade=F)
#no
#devtools::install_github("edwindj/ffbase", subdir="pkg", upgrade=F)
#no
#reticulate::py_install("phate", pip=TRUE)
#no
BiocManager::install("BSgenome.Hsapiens.UCSC.hg38", update=F)#ヒトゲノム(hg38)
BiocManager::install("BSgenome.Hsapiens.NCBI.GRCh38", update=F)#ヒトゲノム(hg38)機能ゲノム学の講義で利用するため2022.05.11にコメントアウトを外した
3. インストール確認
以下を「R コンソール画面」上でコピー&ペースト。
代表的なパッケージ群が正しくインストールされたかを確認しています。
ここの実行結果(特に2回目)で何のエラーメッセージも出なければOK。もし出たら、
「インストール | Rパッケージ | 個別(2018年11月以降)」
を参考にして、エラーが出たパッケージのインストールを個別に行ってください。
library(ape)
library(baySeq)
library(beeswarm)
library(bio3d)
library(biomaRt)
library(Biostrings)
library(bit)
library(BSgenome)
library(BSgenome.Hsapiens.UCSC.hg38)
library(BSgenome.Hsapiens.NCBI.GRCh38)
library(cclust)
library(cluster)
library(clusterStab)
library(corrplot)
library(dada2)
library(data.table)
library(DESeq2)
library(devtools)
library(dplyr)
library(DT)
library(e1071)
library(edgeR)
library(ff)
library(fields)
library(FinePop)
library(FinePop2)
library(FIT)
library(fitdistrplus)
library(GenomicAlignments)
library(GenomicFeatures)
library(GGally)
library(ggplot2)
library(glmnet)
library(GSAR)
library(GSVA)
library(GSVAdata)
library(heatmaply)
library(KernSmooth)
library(knitr)
library(limma)
library(mapdata)
library(maps)
library(MASS)
library(MBCluster.Seq)
library(microseq)
library(msa)
library(MVA)
library(openxlsx)
library(org.Hs.eg.db)
library(pcaMethods)
library(Peptides)
library(phateR)
library(phyloseq)
library(plotly)
library(qqman)
library(qrqc)
library(QuasR)
library(R6)
library(randomForest)
library(RColorBrewer)
library(RCurl)
library(recount)
library(rgl)
library(rmarkdown)
library(rrBLUP)
library(Rsamtools)
library(rentrez)
library(rtracklayer)
library(Rtsne)
library(scatterplot3d)
library(seqinr)
library(seqLogo)
library(shiny)
library(shinydashboard)
library(shinyWidgets)
library(ShortRead)
library(som)
library(splatter)
library(SRAdb)
library(TCC)
library(tidyverse)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
library(BSgenome.Hsapiens.NCBI.GRCh38)
library(umap)
library(zoo)
library(ape)
library(baySeq)
library(beeswarm)
library(bio3d)
library(biomaRt)
library(Biostrings)
library(bit)
library(BSgenome)
library(BSgenome.Hsapiens.UCSC.hg38)
library(cclust)
library(cluster)
library(clusterStab)
library(corrplot)
library(dada2)
library(data.table)
library(DESeq2)
library(devtools)
library(dplyr)
library(DT)
library(e1071)
library(edgeR)
library(ff)
library(fields)
library(FinePop)
library(FinePop2)
library(FIT)
library(fitdistrplus)
library(GenomicAlignments)
library(GenomicFeatures)
library(GGally)
library(ggplot2)
library(glmnet)
library(GSAR)
library(GSVA)
library(GSVAdata)
library(heatmaply)
library(KernSmooth)
library(knitr)
library(limma)
library(mapdata)
library(maps)
library(MASS)
library(MBCluster.Seq)
library(microseq)
library(msa)
library(MVA)
library(openxlsx)
library(org.Hs.eg.db)
library(pcaMethods)
library(phyloseq)
library(Peptides)
library(phateR)
library(plotly)
library(qqman)
library(qrqc)
library(QuasR)
library(R6)
library(randomForest)
library(RColorBrewer)
library(RCurl)
library(recount)
library(rgl)
library(rmarkdown)
library(rrBLUP)
library(Rsamtools)
library(rentrez)
library(rtracklayer)
library(Rtsne)
library(scatterplot3d)
library(seqinr)
library(seqLogo)
library(shiny)
library(shinydashboard)
library(shinyWidgets)
library(ShortRead)
library(som)
library(splatter)
library(SRAdb)
library(TCC)
library(tidyverse)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
library(umap)
library(zoo)
インストール | Rパッケージ | 個別(2018年11月以降)
文字通り、Rのパッケージを個別にインストールするやり方を示します。
このウェブページでは、Bioconductorから提供されているパッケージを数多く利用しています。
2018年10月31日リリースのBioconductor 3.8より、インストール方法が変更されましたのでご注意ください。
具体的には、biocLiteから、BiocManager::installを利用するやり方に変更されました。
「BiocManager::install」は「BiocManagerというパッケージ中にあるinstall関数」という意味です。
intallという関数は他のパッケージでも提供されている可能性があるため、「どのパッケージが提供するinstall関数か」を明示したい場合に玄人がよく利用します。
400MB程度あります...。
param <- "BSgenome.Drerio.UCSC.danRer7"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
param <- "TxDb.Rnorvegicus.UCSC.rn5.refGene"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
param <- "TxDb.Hsapiens.UCSC.hg38.knownGene"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
20MB程度です。
param <- "BSgenome.Celegans.UCSC.ce6"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
param <- "TxDb.Celegans.UCSC.ce6.ensGene"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
20MB程度です。
param <- "BSgenome.Ecoli.NCBI.20080805"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
550MB程度です。
param <- "BSgenome.Cfamiliaris.UCSC.canFam3"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
30MB程度です。
param <- "BSgenome.Dmelanogaster.UCSC.dm2"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
100MB程度です。
param <- "BSgenome.Osativa.MSU.MSU7"
BiocManager::install(param, update=F)
BiocManager::valid(param)
packageVersion(param)
インストール | Rパッケージ | 個別(2018年11月以前)
このウェブページでは、Bioconductorから提供されているパッケージを数多く利用していますが、
2018年10月31日リリースのBioconductor 3.8よりインストール方法が変更されました。
ここでは、2018年11月までの主な「Rパッケージを個別にインストールするやり方」を示します。
400MB程度あります...。
param <- "BSgenome.Drerio.UCSC.danRer7"
source("http://bioconductor.org/biocLite.R")
biocLite(param, suppressUpdates=TRUE)
param <- "TxDb.Rnorvegicus.UCSC.rn5.refGene"
source("http://bioconductor.org/biocLite.R")
biocLite(param, suppressUpdates=TRUE)
基本的な利用法
以下は、インストール | についてを参考にして必要なパッケージのインストールが完了済みのヒトを対象として、
このウェブページの基本的な利用法を簡単に解説したものです。
サンプルデータ
- Illumina/36bp/single-end/human (SRA000299) data (Marioni et al., Genome Res., 2008)
「Kidney 7 samples vs Liver 7 samples」のRNA-seqの遺伝子発現行列データ(SupplementaryTable2.txt)です。
サンプルは二つの濃度(1.5 pM and 3 pM)でシーケンスされており、「3 pMのものが5 samples vs. 5 samples」、「1.5 pMのものが2 samples vs. 2 samples」という構成です。
SupplementaryTable2.txtをエクセルで開くと、7列目以降に発現データがあることがわかります。詳細な情報は以下の通りです(原著論文中のFigure 1からもわかります):
7列目:R1L1Kidney (3 pM)
8列目:R1L2Liver (3 pM)
9列目:R1L3Kidney (3 pM)
10列目:R1L4Liver (3 pM)
11列目:R1L6Liver (3 pM)
12列目:R1L7Kidney (3 pM)
13列目:R1L8Liver (3 pM)
14列目:R2L1Liver (1.5 pM)
15列目:R2L2Kidney (3 pM)
16列目:R2L3Liver (3 pM)
17列目:R2L4Kidney (1.5 pM)
18列目:R2L6Kidney (3 pM)
19列目:R2L7Liver (1.5 pM)
20列目:R2L8Kidney (1.5 pM)
- Illumina/36bp/single-end/human (SRA000299) data (Marioni et al., Genome Res., 2008)
Supplementary table 2のデータを取り扱いやすく加工したデータです。
オリジナルのものは最初の6列が発現データ以外のものだったり、7列目以降も二種類のサンプルが交互に出てくるなど若干R上で表現しずらかったため、以下のようにわかりやすくしたものです。
つまり、サンプルを3pMのものだけにして、「1列目:Genename, 2-6列目:Kidney群, 7-11列目:Liver群」と変更したSupplementaryTable2_changed.txtです:
2列目:R1L1Kidney (3 pM)
3列目:R1L3Kidney (3 pM)
4列目:R1L7Kidney (3 pM)
5列目:R2L2Kidney (3 pM)
6列目:R2L6Kidney (3 pM)
7列目:R1L2Liver (3 pM)
8列目:R1L4Liver (3 pM)
9列目:R1L6Liver (3 pM)
10列目:R1L8Liver (3 pM)
11列目:R2L3Liver (3 pM)
- Illumina/36bp/single-end/human (SRA000299) data (Marioni et al., Genome Res., 2008)
上記SupplementaryTable2_changed.txtをさらに加工したデータ。
NGSデータは(マイクロアレイの黎明期と同じく)金がかかりますので(technical and/or biological) replicatesを簡単には増やせませんので、「1サンプル vs. 1サンプル」比較の局面がまだまだあろうかと思います。
そこで、上記ファイルの2-6列目と7-11列目をそれぞれまとめた(総和をとった)ものSupplementaryTable2_changed2.txtです。
-
カウントデータ(SupplementaryTable2_changed.txt)と長さ情報ファイル(ens_gene_46_length.txt)を読み込んで、
以下を実行して、「配列長情報を含み、カウント数の和が0より大きい行のみ抽出した結果」です。カウントデータファイル(data_marioni.txt)と配列長情報ファイル(length_marioni.txt)。
in_f1 <- "SupplementaryTable2_changed.txt"
in_f2 <- "ens_gene_46_length.txt"
out_f1 <- "data_marioni.txt"
out_f2 <- "length_marioni.txt"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(len)
rownames(len) <- len[,1]
common <- intersect(rownames(len), rownames(data))
data <- data[common,]
len <- len[common,]
dim(data)
dim(len)
head(data)
head(len)
obj <- (rowSums(data) > 0)
data <- data[obj,]
len <- len[obj,]
dim(data)
dim(len)
head(data)
head(len)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
write.table(len, out_f2, sep="\t", append=F, quote=F, row.names=F)
- ABI_SOLiD/25-35bp/single-end/mouse (SRA000306; EB vs. ES) data (Cloonan et al., Nat Methods, 2008)
- Illumina/50bp/paired-end/mouse (SRA012213; liver) data (Robertson et al., Nat Methods, 2010)
- Illumina/35bp/single-end/human (SRA010153; MAQC) data (Bullard et al., BMC Bioinformatics, 2010)
SRR037439から得られるFASTQファイルの最初の2000行分を抽出したMAQC2 brainデータ
非圧縮版:SRR037439.fastq
gzip圧縮版:SRR037439.fastq.gz
-
NBPSeqパッケージ(Di et al., SAGMB, 10:art24, 2011)中の
ArabidopsisのBiological replicatesデータ(G1群3サンプル vs. G2群3サンプル; Cumbie et al., PLoS One, 2011)です。
26,221 genes×6 samplesの「複製あり」タグカウントデータ(data_arab.txt)
オリジナルは"AT4G32850"というIDのものが重複して存在していたため、19520行目のデータを除去してタブ区切りテキストファイルにしています。
- ReCountデータベース(Frazee et al., BMC Bioinformatics, 2011)
マッピング済みの遺伝子発現行列形式のデータセットを多数提供しています。
-
Yeastの二群間比較用データ(2 mutant strains vs. 2 wild-types; technical replicates)
7065行 × 4列のyeast RNA-seqデータ(data_yeast_7065.txt)
yeastRNASeq
(Lee et al., PLoS Genet., 2008)がインストールされていれば、R Console画面上で以下のコマンドのコピペでも取得可能です:
library(yeastRNASeq)
data(geneLevelData)
dim(geneLevelData)
head(geneLevelData)
tmp <- cbind(rownames(geneLevelData), geneLevelData)
write.table(tmp, "data_yeast_7065.txt", sep="\t", append=F, quote=F, row.names=F)
-
上記Yeastの二群間比較用データを用いてGC-content normalizationなどを行う場合に必要な情報
yeast genes (SGD ver. r64)のGC含量(yeastGC_6717.txt)やlength情報(yeastLength_6717.txt)。
EDASeq
(Risso et al., BMC Bioinformatics, 2011)がインストールされていれば、R Console画面上で以下のコマンドのコピペでも取得可能です:
library(EDASeq)
data(yeastGC)
length(yeastGC)
head(yeastGC)
data(yeastLength)
length(yeastLength)
head(yeastLength)
tmp <- cbind(names(yeastGC), yeastGC)
write.table(tmp, "yeastGC_6717.txt", sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(names(yeastLength), yeastLength)
write.table(tmp, "yeastLength_6717.txt", sep="\t", append=F, quote=F, row.names=F)
-
「10.」と「11.」のファイルをもとに共通遺伝子(6685個)のみからなるのサブセットにしたファイル:
data_yeast_common_6685.txt
yeastGC_common_6685.txt
yeastLength_common_6685.txt)
以下のコピペでも得ることができます。
library(yeastRNASeq)
library(EDASeq)
data(geneLevelData)
data(yeastGC)
data(yeastLength)
common <- intersect(rownames(geneLevelData), names(yeastGC))
data <- as.data.frame(geneLevelData[common, ])
GC <- data.frame(GC = yeastGC[common])
length <- data.frame(Length = yeastLength[common])
head(rownames(data))
head(rownames(GC))
head(rownames(length))
tmp <- cbind(rownames(data), data)
write.table(tmp, "data_yeast_common_6685.txt", sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(GC), GC)
write.table(tmp, "yeastGC_common_6685.txt", sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(length), length)
write.table(tmp, "yeastLength_common_6685.txt", sep="\t", append=F, quote=F, row.names=F)
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
10,000 genes×6 samplesの「複製あり」タグカウントデータ(data_hypodata_3vs3.txt)
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3」の計6サンプル分からなります。
全10,000遺伝子中の最初の2,000個(gene_1〜gene_2000まで)が発現変動遺伝子(DEG)です。
全2,000 DEGsの内訳:最初の90%分(gene_1〜gene_1800)がG1群で4倍高発現、残りの10%分(gene_1801〜gene_2000)がG2群で4倍高発現
以下のコピペでも(数値は違ってきますが)同じ条件のシミュレーションデータを作成可能です。:
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.2,
DEG.assign = c(0.9, 0.1),
DEG.foldchange = c(4, 4),
replicates = c(3, 3))
plotFCPseudocolor(tcc)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, "data_hypodata_3vs3.txt", sep="\t", append=F, quote=F, row.names=F)
-
上記のTCCパッケージ中のBiological replicatesを模倣した
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3」の計6サンプルからなるシミュレーションデータから、1列目と4列目のデータを抽出した「複製なし」タグカウントデータ(data_hypodata_1vs1.txt)
よって、「G1_rep1, G2_rep1」の計2サンプル分のみからなります。
以下のコピペでも(数値は違ってきますが)同じ条件のシミュレーションデータを作成可能です。:
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.2,
DEG.assign = c(0.9, 0.1),
DEG.foldchange = c(4, 4),
replicates = c(1, 1))
plotFCPseudocolor(tcc)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, "data_hypodata_1vs1.txt", sep="\t", append=F, quote=F, row.names=F)
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
10,000 genes×9 samplesの「複製あり」タグカウントデータ(data_hypodata_3vs3vs3.txt)
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3, G3_rep1, G3_rep2, G3_rep3」の計9サンプル分からなります。
全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。
全3,000 DEGsの内訳:(1)最初の70%分(gene_1〜gene_2100)がG1群で3倍高発現、(2)次の20%分(gene_2101〜gene_2700)がG2群で10倍高発現、(3)残りの10%分(gene_2701〜gene_3000)がG3群で6倍高発現
以下のコピペでも取得可能です。
out_f <- "data_hypodata_3vs3vs3.txt"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
plotFCPseudocolor(tcc)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
10,000 genes×9 samplesの「複製あり」タグカウントデータ(data_hypodata_2vs4vs3.txt)
「G1_rep1, G1_rep2, G2_rep1, G2_rep2, G2_rep3, G2_rep4, G3_rep1, G3_rep2, G3_rep3」の計9サンプル分からなります。
全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。
全3,000 DEGsの内訳:(1)最初の70%分(gene_1〜gene_2100)がG1群で3倍高発現、(2)次の20%分(gene_2101〜gene_2700)がG2群で10倍高発現、(3)残りの10%分(gene_2701〜gene_3000)がG3群で6倍高発現
以下のコピペでも取得可能です。
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(2, 4, 3))
plotFCPseudocolor(tcc)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, "data_hypodata_2vs4vs3.txt", sep="\t", append=F, quote=F, row.names=F)
- Illumina/35bp/single-end/human (SRA000299; kidney vs. liver) data (Marioni et al., Genome Res., 2008)
-
ランダムな塩基配列から生成したリファレンスゲノム配列データ(ref_genome.fa)。
48, 160, 100, 123, 100 bpの配列長をもつ、計5つの塩基配列を生成しています。
description行は"contig"という記述を基本としています。塩基の存在比はAが28%, Cが22%,
Gが26%, Tが24%にしています。
set.seed関数を利用し、chr3の配列と同じものをchr5としてコピーして作成したのち、2番目と7番目の塩基置換を行っています。
そのため、実際に指定するのは最初の4つ分の配列長のみです。
out_f <- "ref_genome.fa"
param_len_ref <- c(48, 160, 100, 123)
narabi <- c("A","C","G","T")
param_composition <- c(28, 22, 26, 24)
param_desc <- "chr"
param4 <- 3
param5 <- c(2, 7)
library(Biostrings)
enkichikan <- function(fa, p) {
t <- substring(fa, p, p)
t_c <- chartr("CGAT", "GCTA", t)
substring(fa, p, p) <- t_c
return(fa)
}
set.seed(1000)
ACGTset <- rep(narabi, param_composition)
hoge <- NULL
for(i in 1:length(param_len_ref)){
hoge <- c(hoge, paste(sample(ACGTset, param_len_ref[i], replace=T), collapse=""))
}
hoge <- c(hoge, hoge[param4])
hoge[length(param_len_ref)+1] <- enkichikan(hoge[length(param_len_ref)+1], param5[1])
hoge[length(param_len_ref)+1] <- enkichikan(hoge[length(param_len_ref)+1], param5[2])
fasta <- DNAStringSet(hoge)
names(fasta) <- paste(param_desc, 1:length(hoge), sep="")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
-
上記リファレンスゲノム配列データ(ref_genome.fa)に対してbasic alignerでマッピングする際の動作確認用RNA-seqデータ
(sample_RNAseq1.fa)とそのgzip圧縮ファイル(sample_RNAseq1.fa.gz)。
リファレンス配列を読み込んで、list_sub3.txtで与えた部分配列を抽出したものです。
どこに置換を入れているかがわかっているので、basic alignerで許容するミスマッチ数を変えてマップされる or されないの確認ができます。
DNAStringSetオブジェクトを入力として塩基置換を行うDNAString_chartr関数を用いて、最後のリードのみ4番目の塩基にミスマッチを入れています。
in_f1 <- "ref_genome.fa"
in_f2 <- "list_sub3.txt"
out_f <- "sample_RNAseq1.fa"
param <- 4
library(Biostrings)
DNAString_chartr <- function(fa, p) {
str_list <- as.character(fa)
t <- substring(str_list, p, p)
t_c <- chartr("CGAT", "GCTA", t)
substring(str_list, p, p) <- t_c
fa_r <- DNAStringSet(str_list)
names(fa_r) <- names(fa)
return(fa_r)
}
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- NULL
for(i in 1:nrow(posi)){
obj <- names(fasta) == posi[i,1]
hoge <- append(hoge, subseq(fasta[obj], start=posi[i,2], end=posi[i,3]))
}
fasta <- hoge
fasta
fasta[nrow(posi)] <- DNAString_chartr(fasta[nrow(posi)], param)
fasta
description <- paste(posi[,1], posi[,2], posi[,3], sep="_")
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
-
上記リファレンスゲノム配列データ(ref_genome.fa)に対してbasic alignerでマッピングする際の動作確認用RNA-seqデータ(sample_RNAseq2.fa)とそのgzip圧縮ファイル(sample_RNAseq2.fa.gz)。
リファレンス配列を読み込んで、list_sub4.txtで与えた部分配列を抽出したものです。
基本的にジャンクションリードがbasic alignerでマップされず、splice-aware alignerでマップされることを示すために作成したものです。
in_f1 <- "ref_genome.fa"
in_f2 <- "list_sub4.txt"
out_f <- "sample_RNAseq2.fa"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- NULL
hoge_d <- NULL
for(i in 1:nrow(posi)){
uge <- NULL
for(j in 1:(length(posi[i,])/3)){
obj <- names(fasta)==posi[i,3*(j-1)+1]
uge <- paste(uge, subseq(fasta[obj],
start=posi[i,3*(j-1)+2],
end=posi[i,3*(j-1)+3]),
sep="")
}
hoge <- append(hoge, uge)
uge_d <- as.character(posi[i,1])
for(j in 2:(length(posi[i,]))){
uge_d <- paste(uge_d, as.character(posi[i,j]), sep="_")
}
hoge_d <- append(hoge_d, uge_d)
}
fasta <- DNAStringSet(hoge)
names(fasta) <- hoge_d
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
-
GTF (General Transfer Format)形式のヒトサンプルファイル(human_annotation_sub.gtf)です。
EnsemblのFTPサイトから得たヒトの遺伝子アノテーションファイル("Homo_sapiens.GRCh37.73.gtf.gz")を
ここからダウンロードして得て解凍("Homo_sapiens.GRCh37.73.gtf")したのち、
(解凍後のファイルサイズは500MB、2,268,089行×9列のファイルなので)以下のコピペで、ランダムに50,000行分を非復元抽出して得たファイルです。
in_f <- "Homo_sapiens.GRCh37.73.gtf"
out_f <- "human_annotation_sub.gtf"
param <- 50000
data <- read.table(in_f, header=FALSE, sep="\t", quote="")
dim(data)
hoge <- sample(1:nrow(data), param, replace=F)
out <- data[sort(hoge),]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
-
GTF (General Transfer Format)形式のヒトサンプルファイル(human_annotation_sub2.gtf)です。
GTFファイル(human_annotation_sub.gtf)の各行の左端に"chr"を挿入したファイルです。
in_f <- "human_annotation_sub.gtf"
out_f <- "human_annotation_sub2.gtf"
param <- "chr"
data <- read.table(in_f, header=FALSE, sep="\t", quote="")
data[,1] <- paste(param, data[,1], sep="")
write.table(data, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
-
GTF (General Transfer Format)形式のヒトサンプルファイル(human_annotation_sub3.gtf)です。
ヒトゲノム配列("BSgenome.Hsapiens.UCSC.hg19")中の染色体名と一致する遺伝子アノテーション情報のみhuman_annotation_sub2.gtfから抽出したファイルです。
in_f1 <- "human_annotation_sub2.gtf"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f <- "human_annotation_sub3.gtf"
data <- read.table(in_f1, header=FALSE, sep="\t", quote="")
dim(data)
param <- in_f2
library(param, character.only=T)
tmp <- ls(paste("package", param, sep=":"))
hoge <- eval(parse(text=tmp))
keywords <- seqnames(hoge)
keywords
obj <- is.element(as.character(data[,1]), keywords)
out <- data[obj,]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
- Illumina/75bp/single-end/human (SRA061145) data
(Wang et al., Nucleic Acids Res., 2013)
ヒト肺の3群間比較用データ:normal human bronchial epithelial (HBE) cells, human lung cancer A549, and H1299 cells
- Illumina HiSeq 2000/100bp/paired-end/human (GSE42960) data
(Chan et al., Hum. Mol. Genet., 2013)
ヒトPBMCというサンプルの2群間比較用データ:未処理群2サンプル (FRDA05-UT and FRDA19.UTB) vs. ニコチンアミド処理群2サンプル(FRDA05-NicoとFRDA19.NB)。
原著論文中では、GSE42960のみが示されていますが、
日米欧三極のDB(
SRP017580 by SRA;
SRP017580 by DRA;
SRP017580 by ENA)
からも概観できます。
ペアエンドデータのSRR633902_1.fastqを入力として、最初の1000リード分を抽出することで、
SRR633902_1_sub.fastqを作成しています。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR633902_1.fastq.gz"
out_f <- "SRR633902_1_sub.fastq"
param <- 1000
library(ShortRead)
fastq <- readFastq(in_f)
fastq
fastq <- fastq[1:param]
fastq
writeFastq(fastq, out_f, compress=F)
- Illumina Genome Analyzer II/54bp/single-end/human (SRP017142;
GSE42212) data (Neyret-Kahn et al., Genome Res., 2013)
ヒトfibroblastsの2群間比較用データ:3 proliferative samples vs. 3 Ras samples
- Illumina HiSeq 2000 (GPL14844)/50bp/single-end/Rat
(SRP037986; GSE53960) data
(Yu et al., Nat Commun., 2014)
ラットの10組織×雌雄(2種類)×4種類の週齢(2, 6, 21, 104 weeks)×4 biological replicatesの計320サンプルからなるデータ。
- Illumina GAIIx/76bp/paired-end/Drosophila or Illumina HiSeq 2000/100bp/paired-end/Drosophila
(SRP009459; GSE33905) data
(Graveley et al., Nature, 2011; Brown et al., Nature, 2014)
ショウジョウバエの様々な組織のデータ(modENCODE)。29 dissected tissue samplesのstrand-specific, paired-endのbiological replicates (duplicates)だそうです。
- Illumina HiSeq 2000/36bp/single-end/Arabidopsis
(GSE36469) data
(Huang et al., Development, 2012)
シロイヌナズナの2群間比較用データ:4 DEX-treated vs. 4 mock-treated
原著論文中では、GSE36469のみが示されていますが、
日米欧三極のDB(
SRP011435 by SRA;
SRP011435 by DRA;
SRP011435 by ENA)
からも概観できます。
- PacBio/xxx bp/Human
(ERP003225) data
(Sharon et al., Nat Biotechnol., 2013)
ヒトの長鎖RNA-seqデータです。配列長はリードによって異なります。
- PacBio/xxx bp/Chicken
(SRP038897 by DRA; SRP038897 by ENA; SRP038897 by SRA) data
(Sharon et al., PLoS One, 2014)
ニワトリの長鎖RNA-seqデータです。配列長はリードによって異なります。
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample32_ref.fastaとsample32_ngs.fasta)です。
50塩基の長さのリファレンス配列を生成したのち、20塩基長の部分配列を10リード分だけランダム抽出したものです。
塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。
リファレンス配列(仮想ゲノム配列)がsample32_ref.fastaで、
10リードからなる仮想NGSデータがsample32_ngs.fastaです。
リード長20塩基で10リードなのでトータル200塩基となり、50塩基からなる元のゲノム配列の4倍シーケンスしていることになります(4X coverageに相当)。
イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample32_ref.fasta"
out_f2 <- "sample32_ngs.fasta"
param_len_ref <- 50
narabi <- c("A","C","G","T")
param_composition <- c(22, 28, 28, 22)
param_len_ngs <- 20
param_num_ngs <- 10
param_desc <- "kkk"
library(Biostrings)
set.seed(1010)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference <- DNAStringSet(reference)
names(reference) <- param_desc
reference
s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)
s_posi
hoge <- NULL
for(i in 1:length(s_posi)){
hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))
}
fasta <- hoge
description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")
names(fasta) <- description
fasta
writeXStringSet(reference, file=out_f1, format="fasta", width=50)
writeXStringSet(fasta, file=out_f2, format="fasta", width=50)
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample33_ref.fastaとsample33_ngs.fasta)です。
1000塩基の長さのリファレンス配列を生成したのち、20塩基長の部分配列を200リード分だけランダム抽出したものです。
塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。
リファレンス配列(仮想ゲノム配列)がsample33_ref.fastaで、
200リードからなる仮想NGSデータがsample33_ngs.fastaです。
リード長20塩基で200リードなのでトータル4,000塩基となり、1,000塩基からなる元のゲノム配列の4倍シーケンスしていることになります(4X coverageに相当)。
イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
2014年から提供していたものと下記のコピペ実行結果が異なっていることがわかったので、2020年3月16日に中身を変更しました(おそらく昔はset.seedを付けていなかったのだと思われます)。
out_f1 <- "sample33_ref.fasta"
out_f2 <- "sample33_ngs.fasta"
param_len_ref <- 1000
narabi <- c("A","C","G","T")
param_composition <- c(22, 28, 28, 22)
param_len_ngs <- 20
param_num_ngs <- 200
param_desc <- "kkk"
library(Biostrings)
set.seed(1010)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference <- DNAStringSet(reference)
names(reference) <- param_desc
reference
s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)
s_posi
hoge <- NULL
for(i in 1:length(s_posi)){
hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))
}
fasta <- hoge
description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")
names(fasta) <- description
fasta
writeXStringSet(reference, file=out_f1, format="fasta", width=50)
writeXStringSet(fasta, file=out_f2, format="fasta", width=50)
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample34_ref.fastaとsample34_ngs.fasta)です。
1000塩基の長さのリファレンス配列を生成したのち、20塩基長の部分配列を500リード分だけランダム抽出したものです。
塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。
リファレンス配列(仮想ゲノム配列)がsample34_ref.fastaで、
500リードからなる仮想NGSデータがsample34_ngs.fastaです。
リード長20塩基で500リードなのでトータル10,000塩基となり、1,000塩基からなる元のゲノム配列の10倍シーケンスしていることになります(10X coverageに相当)。
イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
2014年から提供していたものと下記のコピペ実行結果が異なっていることがわかったので、2020年3月16日に中身を変更しました(おそらく昔はset.seedを付けていなかったのだと思われます)。
out_f1 <- "sample34_ref.fasta"
out_f2 <- "sample34_ngs.fasta"
param_len_ref <- 1000
narabi <- c("A","C","G","T")
param_composition <- c(22, 28, 28, 22)
param_len_ngs <- 20
param_num_ngs <- 500
param_desc <- "kkk"
library(Biostrings)
set.seed(1010)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference <- DNAStringSet(reference)
names(reference) <- param_desc
reference
s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)
s_posi
hoge <- NULL
for(i in 1:length(s_posi)){
hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))
}
fasta <- hoge
description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")
names(fasta) <- description
fasta
writeXStringSet(reference, file=out_f1, format="fasta", width=50)
writeXStringSet(fasta, file=out_f2, format="fasta", width=50)
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample35_ref.fastaとsample35_ngs.fasta)です。
10000塩基の長さのリファレンス配列を生成したのち、40塩基長の部分配列を2500リード分だけランダム抽出したものです。
塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。
リファレンス配列(仮想ゲノム配列)がsample35_ref.fastaで、
2500リードからなる仮想NGSデータがsample35_ngs.fastaです。
リード長40塩基で2500リードなのでトータル100,000塩基となり、10,000塩基からなる元のゲノム配列の10倍シーケンスしていることになります(10X coverageに相当)。
イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample35_ref.fasta"
out_f2 <- "sample35_ngs.fasta"
param_len_ref <- 10000
narabi <- c("A","C","G","T")
param_composition <- c(22, 28, 28, 22)
param_len_ngs <- 40
param_num_ngs <- 2500
param_desc <- "kkk"
library(Biostrings)
set.seed(1010)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference <- DNAStringSet(reference)
names(reference) <- param_desc
reference
s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)
s_posi
hoge <- NULL
for(i in 1:length(s_posi)){
hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))
}
fasta <- hoge
description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")
names(fasta) <- description
fasta
writeXStringSet(reference, file=out_f1, format="fasta", width=50)
writeXStringSet(fasta, file=out_f2, format="fasta", width=50)
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample36_ref.fastaとsample36_ngs.fasta)です。
10000塩基の長さのリファレンス配列を生成したのち、80塩基長の部分配列を5000リード分だけランダム抽出したものです。
塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。
リファレンス配列(仮想ゲノム配列)がsample36_ref.fastaで、
5,000リードからなる仮想NGSデータがsample36_ngs.fastaです。
リード長80塩基で5,000リードなのでトータル400,000塩基となり、10,000塩基からなる元のゲノム配列の40倍シーケンスしていることになります(40X coverageに相当)。
イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample36_ref.fasta"
out_f2 <- "sample36_ngs.fasta"
param_len_ref <- 10000
narabi <- c("A","C","G","T")
param_composition <- c(22, 28, 28, 22)
param_len_ngs <- 80
param_num_ngs <- 5000
param_desc <- "kkk"
library(Biostrings)
set.seed(1010)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference <- DNAStringSet(reference)
names(reference) <- param_desc
reference
s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)
s_posi
hoge <- NULL
for(i in 1:length(s_posi)){
hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))
}
fasta <- hoge
description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")
names(fasta) <- description
fasta
writeXStringSet(reference, file=out_f1, format="fasta", width=50)
writeXStringSet(fasta, file=out_f2, format="fasta", width=50)
-
k-mer解析用のランダム配列から生成したFASTA形式ファイル(sample37_ref.fastaとsample37_ngs.fasta)です。
10000塩基の長さのリファレンス配列を生成したのち、100塩基長の部分配列を10000リード分だけランダム抽出したものです。
塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。
リファレンス配列(仮想ゲノム配列)がsample37_ref.fastaで、
10,000リードからなる仮想NGSデータがsample37_ngs.fastaです。
リード長100塩基で10,000リードなのでトータル1,000,000塩基となり、10,000塩基からなる元のゲノム配列の100倍シーケンスしていることになります(100X coverageに相当)。
イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成からと基本的に同じです。
out_f1 <- "sample37_ref.fasta"
out_f2 <- "sample37_ngs.fasta"
param_len_ref <- 10000
narabi <- c("A","C","G","T")
param_composition <- c(22, 28, 28, 22)
param_len_ngs <- 100
param_num_ngs <- 10000
param_desc <- "kkk"
library(Biostrings)
set.seed(1010)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference <- DNAStringSet(reference)
names(reference) <- param_desc
reference
s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)
s_posi
hoge <- NULL
for(i in 1:length(s_posi)){
hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))
}
fasta <- hoge
description <- paste(param_desc, s_posi, (s_posi+param_len_ngs-1), sep="_")
names(fasta) <- description
fasta
writeXStringSet(reference, file=out_f1, format="fasta", width=50)
writeXStringSet(fasta, file=out_f2, format="fasta", width=50)
- PacBio/xxx bp/Human
(SRP036136) data
(Tilgner et al., PNAS, 2014)
ヒトの長鎖RNA-seqデータです。配列長はリードによって異なります。
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ
(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル vs. G4群3サンプル vs. G5群3サンプル)です。
10,000 genes×15 samplesの「複製あり」タグカウントデータ(data_hypodata_3vs3vs3vs3vs3.txt)
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3, G3_rep1, G3_rep2, G3_rep3, G4_rep1, G4_rep2, G4_rep3, G5_rep1, G5_rep2, G5_rep3」の計15サンプル分からなります。
全10,000遺伝子(Ngene=10000)中の最初の2,000個(gene_1〜gene_2000まで; 20%なのでPDEG=0.2)が発現変動遺伝子(DEG)です。
全2,000 DEGsの内訳:(1)最初の50%分(gene_1〜gene_1000)がG1群で5倍高発現、
(2)次の20%分(gene_1001〜gene_1400)がG2群で10倍高発現、
(3)次の15%分(gene_1401〜gene_1700)がG3群で8倍高発現、
(4)次の10%分(gene_1701〜gene_1900)がG4群で12倍高発現、
(5)残りの5%分(gene_1901〜gene_2000)がG5群で7倍高発現。
以下のコピペでも取得可能です。
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=10000,PDEG=0.2,
DEG.assign=c(0.5,0.2,0.15,0.1,0.05),
DEG.foldchange=c(5,10,8,12,7),
replicates=c(3, 3, 3, 3, 3))
plotFCPseudocolor(tcc)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, "data_hypodata_3vs3vs3vs3vs3.txt", sep="\t", append=F, quote=F, row.names=F)
-
TCCパッケージ中のBiological replicatesを模倣したシミュレーションデータ
(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
10,000 genes×12 samplesの「複製あり」タグカウントデータ(data_hypodata_4vs4vs4.txt)
「G1_rep1, G1_rep2, G1_rep3, G1_rep4, G2_rep1, G2_rep2, G2_rep3, G2_rep4, G3_rep1, G3_rep2, G3_rep3, G3_rep4」の計12サンプル分からなります。
全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。
全3,000 DEGsの内訳:(1)最初の33.3%分(gene_1〜gene_1000)がG1群で5倍高発現、(2)次の33.3%分(gene_1001〜gene_2000)がG2群で5倍高発現、(3)残りの33.3%分(gene_2001〜gene_3000)がG3群で5倍高発現
以下のコピペでも取得可能です。
out_f <- "data_hypodata_4vs4vs4.txt"
param_replicates <- c(4, 4, 4)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(5, 5, 5)
param_DEGassign <- c(1/3, 1/3, 1/3)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
plotFCPseudocolor(tcc)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
Blekhman et al., Genome Res., 2010のリアルカウントデータです。
Supplementary Table1で提供されているエクセルファイル(http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls; 約4.3MB)
からカウントデータのみ抽出し、きれいに整形しなおしたものがここでの出力ファイルになります。
20,689 genes×36 samplesのカウントデータ(sample_blekhman_36.txt)です。
実験デザインの詳細はFigure S1中に描かれていますが、
ヒト(Homo Sapiens; HS), チンパンジー(Pan troglodytes; PT), アカゲザル(Rhesus macaque; RM)の3種類の生物種の肝臓サンプル(liver sample)の比較を行っています。
生物種ごとにオス3個体メス3個体の計6個体使われており(six individuals; six biological replicates)、
技術的なばらつき(technical variation)を見積もるべく各個体は2つに分割されてデータが取得されています(duplicates; two technical replicates)。
それゆえ、ヒト12サンプル、チンパンジー12サンプル、アカゲザル12サンプルの計36サンプル分のデータということになります。
以下で行っていることはカウントデータの列のみ「ヒトのメス(HSF1, HSF2, HSF3)」, 「ヒトのオス(HSM1, HSM2, HSM3)」,「チンパンジーのメス(PTF1, PTF2, PTF3)」,
「チンパンジーのオス(PTM1, PTM2, PTM3)」, 「アカゲザルのメス(RMF1, RMF2, RMF3)」, 「アカゲザルのオス(RMM1, RMM2, RMM3)」の順番で並び替えたものをファイルに保存しています。
もう少し美しくやることも原理的には可能ですが、そこは本質的な部分ではありませんので、ここではアドホック(その場しのぎ、の意味)な手順で行っています。
当然ながら、エクセルなどでファイルの中身を眺めて完全に列名を把握しているという前提です。
尚、"R1L4.HSF1"と"R4L2.HSF1"が「HSF1というヒトのメス一個体のtechnical replicates」であることは列名や文脈から読み解けます。
#in_f <- "http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls"
in_f <- "suppTable1.xls"
out_f <- "sample_blekhman_36.txt"
hoge <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(hoge)
data <- cbind(
hoge$R1L4.HSF1, hoge$R4L2.HSF1, hoge$R2L7.HSF2, hoge$R3L2.HSF2, hoge$R8L1.HSF3, hoge$R8L2.HSF3,
hoge$R1L1.HSM1, hoge$R5L2.HSM1, hoge$R2L3.HSM2, hoge$R4L8.HSM2, hoge$R3L6.HSM3, hoge$R4L1.HSM3,
hoge$R1L2.PTF1, hoge$R4L4.PTF1, hoge$R2L4.PTF2, hoge$R6L6.PTF2, hoge$R3L7.PTF3, hoge$R5L3.PTF3,
hoge$R1L6.PTM1, hoge$R3L3.PTM1, hoge$R2L8.PTM2, hoge$R4L6.PTM2, hoge$R6L2.PTM3, hoge$R6L4.PTM3,
hoge$R1L7.RMF1, hoge$R5L1.RMF1, hoge$R2L2.RMF2, hoge$R5L8.RMF2, hoge$R3L4.RMF3, hoge$R4L7.RMF3,
hoge$R1L3.RMM1, hoge$R3L8.RMM1, hoge$R2L6.RMM2, hoge$R5L4.RMM2, hoge$R3L1.RMM3, hoge$R4L3.RMM3)
colnames(data) <- c(
"R1L4.HSF1", "R4L2.HSF1", "R2L7.HSF2", "R3L2.HSF2", "R8L1.HSF3", "R8L2.HSF3",
"R1L1.HSM1", "R5L2.HSM1", "R2L3.HSM2", "R4L8.HSM2", "R3L6.HSM3", "R4L1.HSM3",
"R1L2.PTF1", "R4L4.PTF1", "R2L4.PTF2", "R6L6.PTF2", "R3L7.PTF3", "R5L3.PTF3",
"R1L6.PTM1", "R3L3.PTM1", "R2L8.PTM2", "R4L6.PTM2", "R6L2.PTM3", "R6L4.PTM3",
"R1L7.RMF1", "R5L1.RMF1", "R2L2.RMF2", "R5L8.RMF2", "R3L4.RMF3", "R4L7.RMF3",
"R1L3.RMM1", "R3L8.RMM1", "R2L6.RMM2", "R5L4.RMM2", "R3L1.RMM3", "R4L3.RMM3")
rownames(data)<- rownames(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
Blekhman et al., Genome Res., 2010のリアルカウントデータです。
1つ前の例題41とは違って、technical replicatesの2列分のデータは足して1列分のデータとしています。
20,689 genes×18 samplesのカウントデータ(sample_blekhman_18.txt)です。
#in_f <- "http://genome.cshlp.org/content/suppl/2009/12/16/gr.099226.109.DC1/suppTable1.xls"
in_f <- "suppTable1.xls"
out_f <- "sample_blekhman_18.txt"
hoge <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(hoge)
data <- cbind(
hoge$R1L4.HSF1 + hoge$R4L2.HSF1, hoge$R2L7.HSF2 + hoge$R3L2.HSF2, hoge$R8L1.HSF3 + hoge$R8L2.HSF3,
hoge$R1L1.HSM1 + hoge$R5L2.HSM1, hoge$R2L3.HSM2 + hoge$R4L8.HSM2, hoge$R3L6.HSM3 + hoge$R4L1.HSM3,
hoge$R1L2.PTF1 + hoge$R4L4.PTF1, hoge$R2L4.PTF2 + hoge$R6L6.PTF2, hoge$R3L7.PTF3 + hoge$R5L3.PTF3,
hoge$R1L6.PTM1 + hoge$R3L3.PTM1, hoge$R2L8.PTM2 + hoge$R4L6.PTM2, hoge$R6L2.PTM3 + hoge$R6L4.PTM3,
hoge$R1L7.RMF1 + hoge$R5L1.RMF1, hoge$R2L2.RMF2 + hoge$R5L8.RMF2, hoge$R3L4.RMF3 + hoge$R4L7.RMF3,
hoge$R1L3.RMM1 + hoge$R3L8.RMM1, hoge$R2L6.RMM2 + hoge$R5L4.RMM2, hoge$R3L1.RMM3 + hoge$R4L3.RMM3)
colnames(data) <- c(
"HSF1", "HSF2", "HSF3", "HSM1", "HSM2", "HSM3",
"PTF1", "PTF2", "PTF3", "PTM1", "PTM2", "PTM3",
"RMF1", "RMF2", "RMF3", "RMM1", "RMM2", "RMM3")
rownames(data)<- rownames(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
TCCパッケージ中のシミュレーションデータ(G1群1サンプル vs. G2群1サンプル vs. G3群1サンプル)です。
10,000 genes×3 samplesの「複製なし」タグカウントデータ(data_hypodata_1vs1vs1.txt)
「G1_rep1, G2_rep1, G3_rep1」の計3サンプル分からなります。
全10,000遺伝子中の最初の3,000個(gene_1〜gene_3000まで)が発現変動遺伝子(DEG)です。
全3,000 DEGsの内訳:(1)最初の70%分(gene_1〜gene_2100)がG1群で3倍高発現、(2)次の20%分(gene_2101〜gene_2700)がG2群で10倍高発現、
(3)残りの10%分(gene_2701〜gene_3000)がG3群で6倍高発現。
以下のコピペでも取得可能です。
out_f <- "data_hypodata_1vs1vs1.txt"
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(1, 1, 1))
plotFCPseudocolor(tcc)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
- recount2:Collado-Torres et al., Nat Biotechnol., 2017
ReCount(Frazee et al., BMC Bioinformatics, 2011)の後継版です。
Bioconductor上でもrecountというRパッケージが提供されています。
- pasillaパッケージ中の複製あり2群間比較用カウントデータです(孫建強氏 提供情報)。
14,599 genes×7 samplesの「複製あり」タグカウントデータ(sample_pasilla_4vs3.txt)です。
処理前4サンプル(4 untreated) vs. 処理後3サンプル(3 treated)の2群間比較用です。
データの原著論文はBrooks et al., Genome Res., 2011です。
手順としては、pasillaパッケージ中のタブ区切りテキストファイルpasilla_gene_counts.tsvを呼び出し、
それをsample_pasilla_4vs3.txtというファイル名で保存しているだけです。
以下のコピペでも取得可能です。
out_f <- "sample_pasilla_4vs3.txt"
library(pasilla)
hoge <- system.file("extdata",
"pasilla_gene_counts.tsv",
package="pasilla", mustWork=TRUE)
data <- read.csv(hoge, sep="\t", row.names="gene_id")
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
- pasillaパッケージ中の複製なし2群間比較用カウントデータです(孫建強氏 提供情報)。
14,599 genes×2 samplesの「複製なし」タグカウントデータ(sample_pasilla_1vs1.txt)です。
1つ上の例題の4 untreated vs. 3 treatedのオリジナルデータから、1列目と5列目の情報を抽出して、sample_pasilla_1vs1.txtというファイル名で保存しているだけです。
以下のコピペでも取得可能です。
out_f <- "sample_pasilla_1vs1.txt"
param_subset <- c(1, 5)
library(pasilla)
hoge <- system.file("extdata",
"pasilla_gene_counts.tsv",
package="pasilla", mustWork=TRUE)
data <- read.csv(hoge, sep="\t", row.names="gene_id")
data <- data[,param_subset]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
FASTA形式ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa)
に対してbasic alignerでマッピングする際の動作確認用RNA-seqデータ(sample_RNAseq4.fa)。
リファレンス配列を読み込んで、list_sub9.txtで与えた部分配列を抽出したものです。
GFF3形式ののアノテーションファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3)
を用いてマッピング結果からカウント情報を取得する際に、どの領域にマップされたリードがOKなのかを検証するためのリードファイルです。
in_f1 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"
in_f2 <- "list_sub9.txt"
out_f <- "sample_RNAseq4.fa"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- strsplit(names(fasta), " ", fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", 1))
names(fasta) <- hoge2
fasta
hoge <- NULL
for(i in 1:nrow(posi)){
obj <- names(fasta) == posi[i,1]
hoge <- append(hoge, subseq(fasta[obj], start=posi[i,2], end=posi[i,3]))
}
fasta <- hoge
fasta
description <- paste(posi[,1], posi[,2], posi[,3], sep="_")
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
-
MASSパッケージから提供されている
gehanという名前の生存時間解析用データ(sample48.txt;タブ区切りテキストファイル)です。
カプランマイヤー(Kaplan-Meier)法による生存曲線(カプランマイヤー曲線;生存率曲線)作成時の入力ファイルです。
Rと生存時間分析(1)や、
MASSのリファレンスマニュアル56ページ目のgehanの説明部分でも解説されていますが、
これは(ヘッダー行を除く)42行×4列からなる数値行列データです。42行の行数は、42人の白血病患者(leukemia patients)数に相当します。
2人ずつのペアになっており、片方には6-mercaptopurine (6-MP)という薬を投与、もう片方にはプラセボ(control)を投与しています。
行列データの各列には以下に示す情報が格納されています:
1列目(列名:pair)は、患者のid情報が示されています。
例えば1-2行目が1番目のペア、3-4行目が2番目のペアだと読み解きます。
2列目(列名:time)は、寛解時間(単位は週)です。MASSのリファレンスマニュアル56ページ目では、
remission time in weeksと書いてあります。大まかには「元気に過ごせた時間」とか「生存時間」のように解釈しちゃって構いません。
3列目(列名:cens)は、打ち切り(censoring)があったかなかったかという 0 or 1の情報からなります。打ち切りがあったら0、なかったら1です。
このデータの場合は、3列目の0が12個、1が30個です。したがって、12人の患者さんのデータが打ち切りのあるデータ(「上完全データ」と呼ぶそうです)、
30人の患者さんのデータが打ち切りのないデータ(「完全データ」と呼ぶそうです。)ということになります。打ち切りデータというのは、
患者さんとの連絡が取れなくなったなど、何らかの理由で患者さんの状況を把握する手段がなくなったデータのことを指します。
観察期間終了まで生存されている患者さんの場合も、「打ち切りありで0」ということになります。
ちなみに、亡くなったという情報が分かっているデータは打ち切りのないデータに相当します。
4列目(列名:treat)には、プラセボ(control)投与群か6-MP投与群かという「どのような処理を行ったかという処理(treatment)情報」が記載されています。
out_f <- "sample48.txt"
library(MASS)
data(gehan)
tmp <- gehan
head(tmp)
dim(tmp)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
survivalパッケージから提供されている
kidneyという名前の生存時間解析用データ(sample49.txt;タブ区切りテキストファイル)です。
カプランマイヤー(Kaplan-Meier)法による生存曲線(カプランマイヤー曲線;生存率曲線)作成時の入力ファイルです。
survivalのリファレンスマニュアル48ページ目のkidneyの説明部分でも解説されています。
これは(ヘッダー行を除く)76行×7列からなる数値行列データです。
これは、ポータブル透析装置(portable dialysis equipment)を使用している腎臓病患者(kidney patients)向けの、
カテーテル(catheter)挿入時点での感染までの再発時間に関するデータです(McGilchrist and Aisbett, Biometrics, 1991)。
カテーテルは感染以外の理由で除去される場合があります。その場合、観察は打ち切られます。
患者1人につき、2つの観察結果(2 observations)があります。
このデータは76行ありますので、76/2 = 38人分の腎臓病患者のデータがあることになります。
このデータは、生存モデルでのランダム効果(フレイル)を説明するためによく使用されているようです。
行列データの各列には以下に示す情報が格納されています:
1列目(列名:id)は、患者のid情報が示されています。
例えば、(ヘッダー行を除く)最初の1-2行がid = 1の最初の患者さん、次の3-4行がid = 2の患者さん、という風に解釈します。
2列目(列名:time)は、時間です(単位不明)。
3列目(列名:status)は、event statusです。
0 or 1ですが、何が0で何が1かは記載されていません。
Rと生存時間分析(2)
によると、「打ち切りは0、その他は1」となっています。
4列目(列名:age)は、年令情報(in years)です。
5列目(列名:sex)は、性別情報です。1が男性、2が女性です。
6列目(列名:disease)は、「disease type」に関する情報が含まれています。
「0=GN, 1=AN, 2=PKD, 3=Other」だそうです。PKDはほぼ間違いなくpolycystic kidney diseaseの略で、多発性囊胞腎です。
ANは、おそらくacute nephritis (急性腎炎)のこと。GNは、glomerular nephritis (糸球体腎炎)の略なんだろうとは思いますが...。
実際には、数値ではなく「GN or AN or PKD or Other」のいずれかが記載されています。
7列目(列名:frail)は、「frailty estimate from original paper」に関する情報が含まれています。
原著論文(McGilchrist and Aisbett, Biometrics, 1991)から推定した
フレイル(frailty;加齢により心身が老い衰えた状態)の度合いを数値で示したもののようです。数値が大きいほど衰えの度合いが高い?!。
out_f <- "sample49.txt"
library(survival)
data(kidney)
tmp <- kidney
head(tmp)
dim(tmp)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
survivalパッケージから提供されている
colonという名前の生存時間解析用データ(sample50.txt;タブ区切りテキストファイル)です。
カプランマイヤー(Kaplan-Meier)法による生存曲線(カプランマイヤー曲線;生存率曲線)作成時の入力ファイルです。
survivalのリファレンスマニュアル23ページ目のcolonの説明部分でも解説されていますが、
これは(ヘッダー行を除く)1858行×16列からなる数値行列データです。
これは、大腸がんに対する術後補助化学療法(adjuvant chemotherapy)の有効性を示したデータです
(Laurie et al., J Clin Oncol., 1989)。
化学療法としては、低毒性のレバミゾール(Levamisole; 線虫駆虫薬の1種)と、
中程度の毒性のフルオロウラシル(fluorouracil; 5-FU)が使われています。
患者は、何もせずに経過観察(Obsavation)のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類に分けられます。
患者1人につき、2つの記録(two records)があります。再発(recurrence)が1で、死亡(death)が2です。
このデータは1858行ありますので、1858/2 = 929人分の大腸がん患者(colon cancer patients)のデータがあることになります。
それぞれのイベントの種類(event type; etype)ごとに、時間(time)の情報があります。
例えば、idが1の患者さんは、再発(etype = 1)までの時間が968 days、死亡(etype = 2)までの時間が1521 daysだったと解釈します。
データ全体を眺める(特にtime列とetype列を見比べる)とわかりますが、死亡までの時間のほうが再発までの時間よりも短いデータはアリエマセン。
「再発までの時間 <= 死亡までの時間」ということになります。
行列データの各列には以下に示す情報が格納されています:
1列目(列名:id)は、患者のid情報が示されています。
例えば、(ヘッダー行を除く)最初の1-2行がid = 1の最初の患者さん、次の3-4行がid = 2の患者さん、という風に解釈します。
2列目(列名:study)は、全て1であり特に意味はありません。
3列目(列名:rx)は、患者に対してどのような処置(Treatment)を行ったのかという情報が含まれています。
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類です。
4列目(列名:sex)は、性別情報です。0が女性、1が男性です。
5列目(列名:age)は、年令情報(in years)です。
6列目(列名:obstruct)は、「obstruction of colon by tumour」に関する情報が含まれています。
おそらく癌によって腸閉塞が行ったかどうかという0 or 1の情報からなるのだろうと思います。この列をざっと眺めると、0のほうが多いこと、
そしてそれほど腸閉塞の頻度は多くないだろうという素人判断から、0が腸閉塞なし、1が腸閉塞ありなのだろうと思います。
7列目(列名:perfor)は、「perforation of colon」に関する情報が含まれています。
おそらく結腸(大腸のこと)に穴(穿孔)があいちゃったかどうかという0 or 1の情報からなるのだろうと思います。この列をざっと眺めると、0のほうが多いこと、
そしてそれほど穿孔の頻度は多くないだろうという素人判断から、0が穿孔なし、1が穿孔ありなのだろうと思います。
8列目(列名:adhere)は、「adherence to nearby organs」に関する情報が含まれています。
近くの臓器への癒着があったかどうかという0 or 1の情報からなるのだろうと思います。この列をざっと眺めると、0のほうが多いこと、
そしてそれほど癒着の頻度は多くないだろうという素人判断から、0が癒着なし、1が癒着ありなのだろうと思います。
9列目(列名:nodes)は、「number of lymph nodes with detectable cancer」に関する情報が含まれています。
リンパ節への転移に関する情報で、転移のあったリンパ節数(多いほどよくない)です。
10列目(列名:status)は、「censoring status」に関する情報が含まれています。
打ち切り(censoring)があったかなかったかという 0 or 1の情報からなります。打ち切りがあったら0、なかったら1です。
打ち切りデータというのは、患者さんとの連絡が取れなくなったなど、何らかの理由で患者さんの状況を把握する手段がなくなったデータのことを指します。
観察期間終了まで生存されている患者さんの場合は、「打ち切りありで0」ということになります。
ちなみに、亡くなったという情報が分かっているデータは打ち切りのないデータに相当します。
11列目(列名:differ)は、「differentiation of tumour」に関する情報が含まれています。
分化度(differentiation)のことですね。「1=well, 2=moderate, 3=poor」です。高分化型が1、中分化型が2、低分化型が3です。
数値が大きいほど悪性度が高いと解釈します。
12列目(列名:extent)は、「Extent of local spread」に関する情報が含まれています。
「腫瘍の局所的拡大の範囲」と解釈すればよいのでしょうか。「1=submucosa, 2=muscle, 3=serosa, 4=contiguous structures)」です。
大腸粘膜(mucosa)の次の層がsubmucosa、その次が筋層(muscle layer)、その次がserosa (serous membrane;漿膜)、
最後にcontiguous structures (直接隣接する組織)となります。数値が大きいほど深層まで達していると解釈できるので、悪性度が高いと解釈します。
13列目(列名:surg)は、「time from surgery to registration」に関する情報が含まれています。
来訪してから手術までの期間であり、「0=short, 1=long」です。
14列目(列名:node4)は、「more than 4 positive lymph nodes」に関する情報が含まれています。
nodes列が4よりも大きいものが1、4以下が0となっているようですね。
15列目(列名:time)は、「days until event or censoring」に関する情報が含まれています。
イベント(再発 or 死亡)または打ち切り(censoring)までの日数です。数値の大きさは、生存時間の長さを表します。
16列目(列名:etype)は、「event type」に関する情報が含まれています。
再発(recurrence)が1で、死亡(death)が2です。
out_f <- "sample50.txt"
library(survival)
data(colon)
tmp <- colon
head(tmp)
dim(tmp)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
-
MLSeqパッケージ(Goksuluk et al., 2019)から提供されている
cervicalという名前の2群間比較用データ(sample51.txt;タブ区切りテキストファイル)です。
714行×58列からなる数値行列データ(ヘッダー行を除く)です。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
MLSeqパッケージマニュアル
中にも書かれていますが、以下のようなコマンドでも取得可能です。
MLSeqパッケージのインストールが完了していれば、以下のfilepath情報を頼りにしてcervical.txtというファイルを見つければよいだけですが、
filapath情報からたどり着けるのはある程度経験を積んでからというのが現実だと思いますので、わざわざ記載しているのです。
out_f <- "sample51.txt"
library(MLSeq)
filepath <- system.file("extdata/cervical.txt", package="MLSeq")
filepath
data <- read.table(filepath, row.names=1, header=TRUE)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2017
ここの情報は、サブページの「NGSハンズオン講習会2017」に移動しました。
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2016
ここの情報は、サブページの「NGSハンズオン講習会2016」に移動しました。
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGSハンズオン講習会2015
ここの情報は、サブページの「NGSハンズオン講習会2015」に移動しました。
バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ) | NGS速習コース2014
ここの情報は、サブページの「NGS速習コース2014」に移動しました。
イントロ | 一般 | ランダムに行を抽出
例えばタブ区切りテキストファイルのannotation.txtが手元にあり、指定した数の行を(非復元抽出で)ランダムに抽出するやり方を示します。
Linux (UNIX)のgrepコマンドのようなものです。perlのハッシュのようなものです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. タブ区切りテキストファイル(annotation.txt)からランダムに5行分を抽出したい場合:
ヘッダー行はヘッダー行として残す場合のやり方です。
in_f <- "annotation.txt"
out_f <- "hoge1.txt"
param <- 5
data <- read.table(in_f, header=TRUE, sep="\t", quote="")
dim(data)
hoge <- sample(1:nrow(data), param, replace=F)
out <- data[sort(hoge),]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
2. タブ区切りテキストファイル(annotation.txt)からランダムに5行分を抽出したい場合:
ヘッダー行がない場合のやり方です。
in_f <- "annotation.txt"
out_f <- "hoge2.txt"
param <- 5
data <- read.table(in_f, header=FALSE, sep="\t", quote="")
dim(data)
hoge <- sample(1:nrow(data), param, replace=F)
out <- data[sort(hoge),]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
イントロ | 一般 | 任意の文字列を行の最初に挿入
タブ区切りのヒト遺伝子アノテーションファイル(human_annotation_sub.gtf)が手元にあり、
これを入力としてQuasRパッケージを用いてマッピング結果ファイルをもとにカウントデータを得ようと思ってもエラーに遭遇することが多いです。
この原因の一つとして、GFF/GTF形式のアノテーションファイル中の文字列がゲノム情報中の文字列と異なっていることが挙げられます。
例えば、ゲノム配列中の染色体名は"chr1", "chr2", ...などという記述がほとんどですが、これに対応するGFF/GTFの一列目の文字列が"1", "2", ...となっています。
そこでここでは、文字列を一致させるべく、タブ区切りのヒト遺伝子アノテーションファイルの左端に"chr"を挿入するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
ヘッダー行がない場合のやり方です。
in_f <- "human_annotation_sub.gtf"
out_f <- "hoge1.txt"
param <- "chr"
data <- read.table(in_f, header=FALSE, sep="\t", quote="")
data[,1] <- paste(param, data[,1], sep="")
write.table(data, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
イントロ | 一般 | 任意のキーワードを含む行を抽出(基礎)
例えばタブ区切りテキストファイルが手元にあり、この中からリストファイル中の文字列を含む行を抽出するやり方を示します。
Linux (UNIX)のgrepコマンドのようなものであり、perlのハッシュのようなものです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 目的のタブ区切りテキストファイル(annotation.txt)中の第1列目をキーとして、リストファイル(genelist1.txt)中のものが含まれる行全体を出力したい場合:
in_f1 <- "annotation.txt"
in_f2 <- "genelist1.txt"
out_f <- "hoge1.txt"
param <- 1
data <- read.table(in_f1, header=TRUE, sep="\t", quote="")
keywords <- readLines(in_f2)
dim(data)
obj <- is.element(as.character(data[,param]), keywords)
out <- data[obj,]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
2. 目的のタブ区切りテキストファイル(annotation.txt)中の第1列目をキーとして、リストファイル(genelist2.txt)中のものが含まれる行全体を出力したい場合:
in_f1 <- "annotation.txt"
in_f2 <- "genelist2.txt"
out_f <- "hoge2.txt"
param <- 1
data <- read.table(in_f1, header=TRUE, sep="\t", quote="")
keywords <- readLines(in_f2)
obj <- is.element(as.character(data[,param]), keywords)
out <- data[obj,]
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
3. 目的のタブ区切りテキストファイル(annotation.txt)中の第3列目をキーとして、リストファイル(genelist2.txt)中のものが含まれる行全体を出力したい場合:
in_f1 <- "annotation.txt"
in_f2 <- "genelist2.txt"
out_f <- "hoge3.txt"
param <- 3
data <- read.table(in_f1, header=TRUE, sep="\t", quote="")
keywords <- readLines(in_f2)
obj <- is.element(as.character(data[,param]), keywords)
out <- data[obj,]
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
4. 目的のタブ区切りテキストファイル(annotation.txt)に対して、リストファイル(genelist1.txt)中のものが含まれる行全体を出力したい場合:
annotation.txt中にはgene1以外にgene10やgene11もあるため、リストファイル中の文字列(gene1, gene7, gene9)から想定される3行分以外に、
gene10とgene11の行も出力されます。(2016年4月20日追加)
in_f1 <- "annotation.txt"
in_f2 <- "genelist1.txt"
out_f <- "hoge4.txt"
data <- readLines(in_f1)
keywords <- readLines(in_f2)
keywords <- unique(keywords)
hoge <- NULL
for(i in 1:length(keywords)){
hoge <- c(hoge, c(grep(keywords[i], data)))
if(i%%10 == 0) cat(i, "/", length(keywords), "finished\n")
}
hoge <- unique(hoge)
out <- data[hoge]
writeLines(out, out_f)
out_f2 <- "hoge4_hoge.txt"
hoge2 <- NULL
for(i in 1:length(keywords)){
hoge2 <- c(hoge2, list(grep(keywords[i], data)))
}
hoge3 <- sapply(hoge2, paste, collapse="\t")
hoge4 <- paste(keywords, hoge3, sep="\t")
writeLines(hoge4, out_f2)
5. 目的のタブ区切りテキストファイル(annotation.txt)中の第1列目をキーとして、リストファイル(genelist1.txt)中のものに対応するannotation.txt中の第4列目(subcellular_location列)のみを出力する場合:
リストファイルgenelist1.txt中の文字列(gene1, gene7, gene9)が、annotation.txt中の1列目にある行の4列目の情報は、全てnuclearです。(2016年4月20日追加)
in_f1 <- "annotation.txt"
in_f2 <- "genelist1.txt"
out_f <- "hoge5.txt"
param1 <- 1
param2 <- 4
data <- read.table(in_f1, header=TRUE, sep="\t", quote="")
keywords <- readLines(in_f2)
obj <- is.element(as.character(data[,param1]), keywords)
out <- data[obj, param2]
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
6. 例題4と同じことをsapply関数を用いてやる場合:
unique(hoge)のところをunique(unlist(hoge))に変更しました。出力結果は例題4と同じです。(2016年4月20日追加)
in_f1 <- "annotation.txt"
in_f2 <- "genelist1.txt"
out_f <- "hoge6.txt"
data <- readLines(in_f1)
keywords <- readLines(in_f2)
keywords <- unique(keywords)
hoge <- sapply(keywords, grep, x=data)
hoge <- unique(unlist(hoge))
out <- data[hoge]
writeLines(out, out_f)
7. 例題6と同じことを別のファイルを用いてやる場合:
ラットのアノテーション情報ファイル(GPL1355-14795.txt)と、2群間比較で発現変動が確認された遺伝子IDリストファイル(result_rankprod_BAT_id.txt)です。
unique(hoge)のところをunique(unlist(hoge))に変更しました。約15分。(2016年4月20日追加)
in_f1 <- "GPL1355-14795.txt"
in_f2 <- "result_rankprod_BAT_id.txt"
out_f <- "hoge7.txt"
data <- readLines(in_f1)
keywords <- readLines(in_f2)
keywords <- unique(keywords)
hoge <- sapply(keywords, grep, x=data)
hoge <- unique(unlist(hoge))
out <- data[hoge]
writeLines(out, out_f)
8. 例題7と基本的には同じだが、「遺伝子IDリストファイル中の文字列」が「アノテーション情報ファイル中の一番左側」にしか存在しないという前提で高速に探索したい場合:
ラットのアノテーション情報ファイル(GPL1355-14795.txt)と、2群間比較で発現変動が確認された遺伝子IDリストファイル(result_rankprod_BAT_id.txt)です。
unique(hoge)のところをunique(unlist(hoge))に変更しました。約7分。(2016年4月20日追加)
in_f1 <- "GPL1355-14795.txt"
in_f2 <- "result_rankprod_BAT_id.txt"
out_f <- "hoge8.txt"
data <- readLines(in_f1)
keywords <- readLines(in_f2)
keywords <- unique(keywords)
hoge <- sapply(paste("^", keywords, sep=""), grep, x=data)
hoge <- unique(unlist(hoge))
out <- data[hoge]
writeLines(out, out_f)
9. 8を基本として、8の出力ファイルは対象の行の情報全てを出力するものであったが、13列目のRefSeq Transcript IDに相当するもののみ抽出したい場合:
ラットのアノテーション情報ファイル(GPL1355-14795.txt), 二群間比較で発現変動が確認された遺伝子IDリストファイル(result_rankprod_BAT_id.txt)
in_f1 <- "GPL1355-14795.txt"
in_f2 <- "result_rankprod_BAT_id.txt"
out_f <- "hoge9.txt"
param <- 13
data <- readLines(in_f1)
keywords <- readLines(in_f2)
keywords <- unique(keywords)
hoge <- sapply(paste("^", keywords, sep=""), grep, x=data)
hoge <- unique(hoge)
hoge2 <- data[hoge]
hoge3 <- strsplit(hoge2, "\t")
out <- unlist(lapply(hoge3, "[[", param))
writeLines(out, out_f)
10. 9を基本として、8の出力ファイルは対象の行の情報全てを出力するものであったが、13列目のRefSeq Transcript IDに相当するもののみ抽出したい場合:
ラットのアノテーション情報ファイル(GPL1355-14795.txt), 二群間比較で発現変動が確認された遺伝子IDリストファイル(result_rankprod_BAT_id.txt)
アノテーション情報ファイルの形式は"#"から始まる行以外は同じ列数なので、行列形式などにすることが可能なことを利用している(9に比べて一般性は劣るがより劇的に早い計算が可能)
in_f1 <- "GPL1355-14795.txt"
in_f2 <- "result_rankprod_BAT_id.txt"
out_f <- "hoge10.txt"
param <- 13
data <- readLines(in_f1)
keywords <- readLines(in_f2)
keywords <- unique(keywords)
hoge <- grep("^#", data)
data <- data[-hoge]
hoge1 <- strsplit(data, "\t")
hoge2 <- unlist(lapply(hoge1, "[[", param))
names(hoge2) <- unlist(lapply(hoge1, "[[", 1))
hoge3 <- hoge2[keywords]
out <- unique(hoge3)
writeLines(out, out_f)
11. 10を基本として、遺伝子IDリストに対応するRefSeq Transcript IDを抽出ところまでは同じだが、RefSeq IDが同じで遺伝子IDリストにないもの(common)も存在するのでその分を考慮:
ラットのアノテーション情報ファイル(GPL1355-14795.txt), 二群間比較で発現変動が確認された遺伝子IDリストファイル(result_rankprod_BAT_id.txt)
アノテーション情報ファイルの形式は"#"から始まる行以外は同じ列数なので、行列形式などにすることが可能なことを利用している(9に比べて一般性は劣るがより劇的に早い計算が可能)
in_f1 <- "GPL1355-14795.txt"
in_f2 <- "result_rankprod_BAT_id.txt"
out_f1 <- "result_rankprod_BAT_RefSeq_DEG.txt"
out_f2 <- "result_rankprod_BAT_RefSeq_nonDEG.txt"
param <- 13
data <- readLines(in_f1)
geneid_DEG <- readLines(in_f2)
hoge <- grep("^#", data)
data <- data[-hoge]
hoge1 <- strsplit(data, "\t")
hoge2 <- unlist(lapply(hoge1, "[[", param))
names(hoge2) <- unlist(lapply(hoge1, "[[", 1))
tmp_DEG <- unique(hoge2[geneid_DEG])
geneid_nonDEG <- setdiff(names(hoge2), geneid_DEG)
tmp_nonDEG <- unique(hoge2[geneid_nonDEG])
common <- intersect(tmp_DEG, tmp_nonDEG)
out_DEG <- setdiff(tmp_DEG, common)
out_nonDEG <- setdiff(tmp_nonDEG, common)
writeLines(out_DEG, out_f1)
writeLines(out_nonDEG, out_f2)
12. 目的のタブ区切りテキストファイル(annotation.txt)中の第1列目をキーとして、param2で指定した文字列が含まれる行全体を出力したい場合:
in_f <- "annotation.txt"
out_f <- "hoge12.txt"
param1 <- 1
param2 <- c("gene1", "gene7", "gene9")
data <- read.table(in_f, header=TRUE, sep="\t", quote="")
dim(data)
obj <- is.element(as.character(data[,param1]), param2)
out <- data[obj,]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
13. 目的のタブ区切りテキストファイル(annotation2.txt)中の第1列目をキーとして、param2で指定した文字列が含まれる行全体を出力したい場合:
入力ファイル中にヘッダー行がない場合の読み込み例です。
in_f <- "annotation2.txt"
out_f <- "hoge13.txt"
param1 <- 1
param2 <- c("gene1", "gene7", "gene9")
data <- read.table(in_f, header=F, sep="\t", quote="")
dim(data)
obj <- is.element(as.character(data[,param1]), param2)
out <- data[obj,]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
乳酸菌ゲノム(Lactobacillus casei 12A)のアノテーションファイルです。
4.をベースに作成。unique(hoge)のところをunique(unlist(hoge))に変更しました。(2016年4月20日追加)
in_f <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f <- "hoge14.txt"
param <- "ID=gene"
data <- readLines(in_f)
length(data)
hoge <- sapply(param, grep, x=data)
hoge <- unique(unlist(hoge))
out <- data[hoge]
length(out)
writeLines(out, out_f)
15. GFF3形式ファイル(annotation.gff)に対して、"CDS"という文字列が含まれる行全体を出力したい場合:
2019年5月13日の講義で利用したファイルです。
in_f <- "annotation.gff"
out_f <- "hoge15.txt"
param <- "CDS"
data <- readLines(in_f)
length(data)
hoge <- sapply(param, grep, x=data)
hoge <- unique(unlist(hoge))
out <- data[hoge]
length(out)
writeLines(out, out_f)
イントロ | 一般 | ランダムな塩基配列を生成
タイトル通り、「任意の長さ」で「任意の塩基組成」からなるつランダムな塩基配列を生成するやり方を示します。
A,C,G,Tの数値を指定することで任意の塩基組成にできるようになっています。指定する数値の合計は別に100にならなくてもかまいません。
例えば「全てを1にしておけば、四種類の塩基の出現確率の期待値が25%」になりますし、「A=0, C=705, G=89, T=206みたいな指定法だと、
(数値の合計が1000なので)塩基Cの出現確率が70.5%」みたいなこともできます。
1. 50塩基の長さのランダムな塩基配列を生成する場合:
塩基の存在比はAが20%, Cが30%, Gが30%, Tが20%にしています。
param_len_ref <- 50
narabi <- c("A","C","G","T")
param_composition <- c(20, 30, 30, 20)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference
2. 塩基配列を生成させて任意のdescription("kkk")を追加してFASTA形式ファイルで保存したい場合:
70塩基の長さのランダムな塩基配列を生成するやり方です。
塩基の存在比はAが23%, Cが27%, Gが28%, Tが22%にしています。
out_f <- "hoge2.fasta"
param_len_ref <- 70
narabi <- c("A","C","G","T")
param_composition <- c(23, 27, 28, 22)
param_desc <- "kkk"
library(Biostrings)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference
fasta <- DNAStringSet(reference)
names(fasta) <- param_desc
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. 任意の配列長をもつものを複数個作ってmulti-FASTAファイルとして保存したい場合:
24, 103, 65の配列長をもつ、計3つの塩基配列を生成しています。
description行は"contig"という記述を基本としています。
out_f <- "hoge3.fasta"
param_len_ref <- c(24, 103, 65)
narabi <- c("A","C","G","T")
param_composition <- c(20, 30, 30, 20)
param_desc <- "contig"
library(Biostrings)
ACGTset <- rep(narabi, param_composition)
hoge <- NULL
for(i in 1:length(param_len_ref)){
hoge <- c(hoge, paste(sample(ACGTset, param_len_ref[i], replace=T), collapse=""))
}
fasta <- DNAStringSet(hoge)
names(fasta) <- paste(param_desc, 1:length(hoge), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. 配列長情報を含むファイル(seq_length.txt; 中身は「24, 103, 65, 49」という4行からなる数値情報)を読み込む場合:
塩基の存在比はAが26%, Cが27%,
Gが24%, Tが23%にしています。
in_f <- "seq_length.txt"
out_f <- "hoge4.fasta"
narabi <- c("A","C","G","T")
param_composition <- c(26, 27, 24, 23)
param_desc <- "contig"
library(Biostrings)
param_len_ref <- readLines(in_f)
ACGTset <- rep(narabi, param_composition)
hoge <- NULL
for(i in 1:length(param_len_ref)){
hoge <- c(hoge, paste(sample(ACGTset, param_len_ref[i], replace=T), collapse=""))
}
fasta <- DNAStringSet(hoge)
names(fasta) <- paste(param_desc, 1:length(hoge), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. 同一パラメータを与えたときには常に同じ塩基配列が生成されるようにしたい場合:
48, 160, 100, 123の配列長をもつ、計4つの塩基配列を生成しています。
description行は"contig"という記述を基本としています。
塩基の存在比はAが28%, Cが22%,
Gが26%, Tが24%にしています。
set.seed関数を追加しているだけです。
out_f <- "hoge5.fasta"
param_len_ref <- c(48, 160, 100, 123)
narabi <- c("A","C","G","T")
param_composition <- c(28, 22, 26, 24)
param_desc <- "chr"
library(Biostrings)
set.seed(1000)
ACGTset <- rep(narabi, param_composition)
hoge <- NULL
for(i in 1:length(param_len_ref)){
hoge <- c(hoge, paste(sample(ACGTset, param_len_ref[i], replace=T), collapse=""))
}
fasta <- DNAStringSet(hoge)
names(fasta) <- paste(param_desc, 1:length(hoge), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
6. 同一パラメータを与えたときには常に同じ塩基配列が生成されるようにしたい場合2:
48, 160, 100, 123の配列長をもつ、計4つの塩基配列を生成しています。
description行は"contig"という記述を基本としています。
塩基の存在比はAが28%, Cが22%,
Gが26%, Tが24%にしています。
set.seed関数を追加し、chr3の配列と同じものをchr5としてコピーして作成したのち、一部の塩基置換を行っています。
out_f <- "hoge6.fasta"
param_len_ref <- c(48, 160, 100, 123)
narabi <- c("A","C","G","T")
param_composition <- c(28, 22, 26, 24)
param_desc <- "chr"
library(Biostrings)
enkichikan <- function(fa, p) {
t <- substring(fa, p, p)
t_c <- chartr("CGAT", "GCTA", t)
substring(fa, p, p) <- t_c
return(fa)
}
set.seed(1000)
ACGTset <- rep(narabi, param_composition)
hoge <- NULL
for(i in 1:length(param_len_ref)){
hoge <- c(hoge, paste(sample(ACGTset, param_len_ref[i], replace=T), collapse=""))
}
hoge <- c(hoge, hoge[3])
hoge[5] <- enkichikan(hoge[5], 2)
hoge[5] <- enkichikan(hoge[5], 7)
fasta <- DNAStringSet(hoge)
names(fasta) <- paste(param3, 1:length(hoge), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 任意の長さの可能な全ての塩基配列を作成
Biostringsパッケージを用いて"A", "C", "G", "T"からなる
任意の長さのk塩基(k-mer)からなる全ての塩基配列を作成するやり方を示します。
「ファイル」−「ディレクトリの変更」で出力結果ファイルを保存したいディレクトリに移動し以下をコピペ。
1. k=3として、4k = 43 = 64通りの3塩基からなる可能な配列を作成したい場合:
out_f <- "hoge1.txt"
param_kmer <- 3
library(Biostrings)
out <- mkAllStrings(c("A", "C", "G", "T"), param_kmer)
writeLines(out, out_f)
2. k=5として、4k = 45 = 1024通りの5塩基からなる可能な配列を作成したい場合:
out_f <- "hoge2.txt"
param_kmer <- 5
library(Biostrings)
out <- mkAllStrings(c("A", "C", "G", "T"), param_kmer)
writeLines(out, out_f)
イントロ | 一般 | 任意の位置の塩基を置換
任意の位置の塩基を置換するやり方を示します。ベタな書き方ですがとりあえず。。。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample1.fasta"
out_f <- "hoge1.fasta"
param1 <- 5
param2 <- "G"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- as.character(fasta)
substring(hoge, param1, param1) <- param2
hoge <- DNAStringSet(hoge)
names(hoge) <- names(fasta)
fasta <- hoge
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
in_f <- "sample2.fasta"
out_f <- "hoge2.fasta"
param1 <- 5
param2 <- "G"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- as.character(fasta)
substring(hoge, param1, param1) <- param2
hoge <- DNAStringSet(hoge)
names(hoge) <- names(fasta)
fasta <- hoge
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
param1で指定した位置の塩基を相補鎖(C -> G, G -> C, A -> T, T -> A)に置換するやり方です。そのような関数を作成して実行しています。
in_f <- "sample2.fasta"
out_f <- "hoge3.fasta"
param1 <- 5
library(Biostrings)
DNAString_chartr <- function(fa, p) {
str_list <- as.character(fa)
t <- substring(str_list, p, p)
t_c <- chartr("CGAT", "GCTA", t)
substring(str_list, p, p) <- t_c
fa_r <- DNAStringSet(str_list)
names(fa_r) <- names(fa)
return(fa_r)
}
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- DNAString_chartr(fasta, param1)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 指定した範囲の配列を取得 | Biostrings
Biostringsパッケージ中のsubseq関数を用いて、
single-FASTA形式やmulti-FASTA形式ファイルから様々な部分配列を取得するやり方を示します。
この項目は、「この染色体の、ここから、ここまで」という指定の仕方になります。
例えば入力ファイルがヒトゲノムだった場合に、chr3の20000から500000の座標の配列取得を行いたい場合などに利用します。
したがって、chr4とchr8の配列のみ抽出といったやり方には対応していませんのでご注意ください。
また、ファイルダウンロード時に、*.fastaという拡張子が*.txtに勝手に変更されることがありますのでご注意ください。
ここでは、
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. (single-)FASTA形式ファイル(sample1.fasta)の場合:
任意の範囲 (始点が3, 終点が9)の配列を抽出するやり方です。
in_f <- "sample1.fasta"
out_f <- "hoge1.fasta"
param <- c(3, 9)
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- subseq(fasta, param[1], param[2])
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. RefSeqのhuman mRNAのmulti-FASTA形式のファイル (h_rna.fasta)の場合:
任意のRefSeq ID (例:NM_203348.1)の任意の範囲 (例:始点が2, 終点が5)の配列の抽出を行うやり方です。
in_f <- "h_rna.fasta"
out_f <- "hoge2.fasta"
param1 <- "NM_203348.1"
param2 <- c(2, 5)
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(names(fasta) == param1)
fasta <- subseq(fasta[obj], param2[1], param2[2])
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. RefSeqのhuman mRNAのmulti-FASTA形式のファイル (h_rna.fasta)の場合:
目的のaccession番号が複数ある場合に対応したものです。
予め用意しておいた「1列目:accession, 2列目:start位置, 3列目:end位置」からなるリストファイル (list_sub1.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
in_f1 <- "h_rna.fasta"
in_f2 <- "list_sub1.txt"
out_f <- "hoge3.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- NULL
for(i in 1:nrow(posi)){
obj <- names(fasta) == posi[i,1]
hoge <- append(hoge, subseq(fasta[obj], start=posi[i,2], end=posi[i,3]))
}
fasta <- hoge
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
目的のaccession番号が複数ある場合に対応したものです。
予め用意しておいた「1列目:accession, 2列目:start位置, 3列目:end位置」からなるリストファイル (list_sub2.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
in_f1 <- "hoge4.fa"
in_f2 <- "list_sub2.txt"
out_f <- "hoge4.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- NULL
for(i in 1:nrow(posi)){
obj <- names(fasta) == posi[i,1]
hoge <- append(hoge, subseq(fasta[obj], start=posi[i,2], end=posi[i,3]))
}
fasta <- hoge
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. multi-FASTA形式のファイル (ref_genome.fa)ファイルの場合:
目的のaccession番号が複数ある場合に対応したものです。
予め用意しておいた「1列目:accession, 2列目:start位置, 3列目:end位置」からなるリストファイル (list_sub3.txt)
を読み込ませて、目的の部分配列のmulti-FASTAファイルをゲットするやり方です。
in_f1 <- "ref_genome.fa"
in_f2 <- "list_sub3.txt"
out_f <- "hoge5.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- NULL
for(i in 1:nrow(posi)){
obj <- names(fasta) == posi[i,1]
tmp <- subseq(fasta[obj], start=posi[i,2], end=posi[i,3])
hoge <- append(hoge, tmp)
}
fasta <- hoge
fasta
description <- paste(posi[,1], posi[,2], posi[,3], sep="_")
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
6. multi-FASTA形式のファイル (genome.fna)ファイルの場合:
例題5と基本的に同じで、入力ファイルが異なるだけです。
予め用意しておいた「1列目:accession, 2列目:start位置, 3列目:end位置」からなるリストファイル (list_20190513.txt)
を読み込ませて、2,311個のCDSからなるmulti-FASTAファイルをゲットするやり方です。
in_f1 <- "genome.fna"
in_f2 <- "list_20190513.txt"
out_f <- "hoge6.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- NULL
for(i in 1:nrow(posi)){
obj <- names(fasta) == posi[i,1]
tmp <- subseq(fasta[obj], start=posi[i,2], end=posi[i,3])
hoge <- append(hoge, tmp)
}
fasta <- hoge
fasta
description <- paste(posi[,1], posi[,2], posi[,3], sep="_")
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
7. multi-FASTA形式のファイル (genome.fna)ファイルの場合:
例題6と基本的に同じですが、4列目にストランド情報を含むリストファイル (list_20190513_strand.txt)
を読み込ませて、ストランドを適切に反映させた2,311個のCDSからなるmulti-FASTAファイルをゲットするやり方です。
in_f1 <- "genome.fna"
in_f2 <- "list_20190513_strand.txt"
out_f <- "hoge7.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
posi <- read.table(in_f2)
fasta
hoge <- NULL
for(i in 1:nrow(posi)){
obj <- names(fasta) == posi[i,1]
tmp <- subseq(fasta[obj], start=posi[i,2], end=posi[i,3])
if(posi[i,4] == "-"){tmp <- reverseComplement(tmp)}
hoge <- append(hoge, tmp)
}
fasta <- hoge
fasta
description <- paste(posi[,1], posi[,2], posi[,3], sep="_")
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 指定したID(染色体やdescription)の配列を取得
multi-FASTA形式ファイルからリストファイルで指定したID (description行の記載内容と全く同じ染色体名などの配列ID)の配列を取得するやり方を示します。
例えば入力ファイルがヒトゲノムだった場合に、chr4とchr8の配列のみ抽出したい場合などに利用します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. RefSeqのhuman mRNAのmulti-FASTA形式のファイル (h_rna.fasta)の場合:
任意のRefSeq ID NM_203348.1の配列抽出を行うやり方です。
ファイルダウンロード時に、*.fastaという拡張子が*.txtに勝手に変更されることがありますのでご注意ください。
in_f <- "h_rna.fasta"
out_f <- "hoge1.fasta"
param1 <- "NM_203348.1"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(names(fasta) == param1)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. RefSeqのhuman mRNAのmulti-FASTA形式のファイル (h_rna.fasta)の場合:
うまくいかない例です。NM_203348.1は確かに入力ファイル中に存在するが、
バージョン情報を除いたNM_203348だとうまくいきません。
理由は、両者の文字列は完全に同じというわけではないからです。
in_f <- "h_rna.fasta"
out_f <- "hoge2.fasta"
param1 <- "NM_203348"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(names(fasta) == param1)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. RefSeqのhuman mRNAのmulti-FASTA形式のファイル (h_rna.fasta)の場合:
うまくいかない例です。"NM_203348.1"は確かに入力ファイル中に存在するが、
スペースが余分に含まれる"NM_203348.1 "だとうまくいきません。
理由は、両者の文字列は完全に同じというわけではないからです。
in_f <- "h_rna.fasta"
out_f <- "hoge3.fasta"
param1 <- "NM_203348.1 "
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(names(fasta) == param1)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. RefSeqのhuman mRNAのmulti-FASTA形式のファイル (h_rna.fasta)の場合:
目的のIDが複数ある場合に対応したものです。予め用意しておいたリストファイル (list_sub5.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
in_f1 <- "h_rna.fasta"
in_f2 <- "list_sub5.txt"
out_f <- "hoge4.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. RefSeqのhuman mRNAのmulti-FASTA形式のファイル (h_rna.fasta)の場合:
目的のIDが複数ある場合に対応したものです。予め用意しておいたリストファイル (list_sub6.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
バージョン番号やスペースを含むIDは抽出できないという例です。
リスト中の3つのうち、最後のIDの配列のみ抽出できていることがわかります。
in_f1 <- "h_rna.fasta"
in_f2 <- "list_sub6.txt"
out_f <- "hoge5.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
目的のIDが複数ある場合に対応したものです。予め用意しておいたリストファイル (list_sub7.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
"contig_2"の配列は取得できていますが、"contig_4"の配列は取得できていないことがわかります。
理由は、"contig_4 "とスペースが入っているためです。
in_f1 <- "hoge4.fa"
in_f2 <- "list_sub7.txt"
out_f <- "hoge6.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
7. multi-FASTA形式のファイル (ref_genome.fa)ファイルの場合:
目的のIDが複数ある場合に対応したものです。予め用意しておいたリストファイル (list_sub8.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
指定したつもりの4つの染色体番号のうち、"chr1 "はスペースのため、そして"chr_5"は余分な文字の挿入のため抽出できていないことがわかります。
in_f1 <- "ref_genome.fa"
in_f2 <- "list_sub8.txt"
out_f <- "hoge7.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
目的のIDが複数ある場合に対応したものです。予め用意しておいたリストファイル (list_sub8.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
指定したつもりの4つの染色体番号のうち、"chr1 "はスペースのため、そして"chr_5"は余分な文字の挿入のため抽出できていないことがわかります。
multi-FASTAの入力ファイルは、chr4という全く同じ配列が重複して存在しますが、それがそのまま反映されていることが分かります。
in_f1 <- "ref_genome_redun.fa"
in_f2 <- "list_sub8.txt"
out_f <- "hoge8.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
目的のIDが複数ある場合に対応したものです。予め用意しておいたリストファイル (list_sub8.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
指定したつもりの4つの染色体番号のうち、"chr1 "はスペースのため、そして"chr_5"は余分な文字の挿入のため抽出できていないことがわかります。
multi-FASTAの入力ファイルは、chr4という全く同じ配列が重複して存在します。
それがそのまま反映されるのが嫌で、重複を除きたい(non-redundantにしたい)場合があります。それに対応したやり方です。
in_f1 <- "ref_genome_redun.fa"
in_f2 <- "list_sub8.txt"
out_f <- "hoge9.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
fasta <- unique(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
目的のIDが複数ある場合に対応したものです。予め用意しておいたリストファイル (list_sub8.txt)
を読み込ませて、目的の配列のmulti-FASTAファイルをゲットするやり方です。
指定したつもりの4つの染色体番号のうち、"chr1 "はスペースのため、そして"chr_5"は余分な文字の挿入のため抽出できていないことがわかります。
multi-FASTAの入力ファイルは、chr4という全く同じ配列が重複して存在します。
それがそのまま反映されるのが嫌で、重複を除きたい(non-redundantにしたい)場合があります。それに対応したやり方です。
FASTA形式ファイルの読み込み部分で、Biostringsパッケージ中のreadDNAStringSet関数ではなく、seqinrパッケージ中のread.fasta関数を用いるやり方です。
in_f1 <- "ref_genome_redun.fa"
in_f2 <- "list_sub8.txt"
out_f <- "hoge10.fasta"
library(Biostrings)
library(seqinr)
hoge <- read.fasta(in_f, seqtype="DNA", as.string=TRUE)
fasta <- DNAStringSet(as.character(hoge))
names(fasta) <- names(hoge)
keywords <- readLines(in_f2)
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
fasta <- unique(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 翻訳配列(translate)を取得 | について
塩基配列を入力として、アミノ酸配列を取得するプログラムです。
Galaxyでもできるはずです。
イントロ | 一般 | 翻訳配列(translate)を取得 | Biostrings
Biostringsパッケージを用いて塩基配列を読み込んでアミノ酸配列に翻訳するやり方を示します。
翻訳のための遺伝コード(genetic code)は、Standard Genetic Codeだそうです。
もちろん生物種?!によって多少違い(variants)があるようで、"Standard", "SGC0", "Vertebrate Mitochondrial", "SGC1"などいろいろ選べるようです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
multi-FASTAではないsingle-FASTA形式ファイルです。
in_f <- "sample1.fasta"
out_f <- "hoge1.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- translate(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. (multi-)FASTA形式ファイル(sample4.fasta)の場合:
配列中にACGT以外のものが存在するためエラーが出る例です。
4番目の配列(つまりgene_4)の17番目のポジションがNなので妥当です。
in_f <- "sample4.fasta"
out_f <- "hoge2.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- translate(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. (multi-)FASTA形式ファイル(sample4.fasta)の場合:
エラーへの対策として、ACGTのみからなる配列を抽出したサブセットを抽出しています。
翻訳はそれらのサブセットのみに対して行っているので「文字が塩基ではない」という類のエラーがなくなっていることがわかります。
出力ファイル中の*は終始コドン(stop codon)を表すようですね。
in_f <- "sample4.fasta"
out_f <- "hoge3.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(fasta)[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(fasta)[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
fasta <- translate(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. multi-FASTA形式ファイル(h_rna.fasta)の場合:
配列中にNを含むものが出現したところでエラーが出て止まる例です。
「以下にエラー .Call2("DNAStringSet_translate", x, skip_code, dna_codes[codon_alphabet], :
in 'x[[406]]': not a base at pos 498」といったエラーが見られると思います。
これは406番目の配列の498番目のポジションの文字が塩基ではないと文句を言っています。
in_f <- "h_rna.fasta"
out_f <- "hoge4.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- translate(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. multi-FASTA形式ファイル(h_rna.fasta)の場合:
エラーへの対策として、ACGTのみからなる配列を抽出したサブセットを抽出しています。
翻訳はそれらのサブセットのみに対して行っているのでエラーは出なくなっていることがわかります。
in_f <- "h_rna.fasta"
out_f <- "hoge5.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(fasta)[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(fasta)[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
fasta <- translate(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
6. multi-FASTA形式ファイル(h_rna.fasta)の場合:
5.と基本的に同じです。translate関数実行時に、genetic.codeオプションのデフォルトを明記しています。
in_f <- "h_rna.fasta"
out_f <- "hoge6.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- rowSums(alphabetFrequency(fasta)[,1:4])
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
fasta <- translate(fasta, genetic.code=GENETIC_CODE)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
7. multi-FASTA形式ファイル(h_rna.fasta)の場合:
translate関数実行時に、genetic.codeオプションをgetGeneticCode("SGC1")に変更しています。
遺伝コードが変わっているので、6.と異なった結果となります。
in_f <- "h_rna.fasta"
out_f <- "hoge7.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(fasta)[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(fasta)[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
fasta <- translate(fasta, genetic.code=getGeneticCode("SGC1"))
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 翻訳配列(translate)を取得 | seqinr(Charif_2005)
seqinrパッケージを用いて塩基配列を読み込んでアミノ酸配列に翻訳するやり方を示します。
本気で翻訳配列を取得する場合にはこちらの利用をお勧めします。翻訳できないコドンはアミノ酸X(不明なアミノ酸)に変換してくれたり、
translate関数のオプションとしてambiguous=Tとすると、翻訳できるものは可能な限り翻訳してくれます(高橋 広夫 氏提供情報)。
lapply関数を用いるやり方(高橋 広夫 氏提供情報)とsapply関数を用いるやり方(甲斐 政親 氏提供情報)を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
multi-FASTAではないsingle-FASTA形式ファイルです。
in_f <- "sample1.fasta"
out_f <- "hoge1.fasta"
library(seqinr)
hoge <- read.fasta(in_f, seqtype="DNA")
hoge
hoge <- lapply(hoge, function(x){
translate(x, ambiguous=T)
})
hoge
write.fasta(hoge, names=names(hoge), file.out=out_f, nbchar=50)
2. (multi-)FASTA形式ファイル(sample4.fasta)の場合:
lapply関数を用いるやり方です。
in_f <- "sample4.fasta"
out_f <- "hoge2.fasta"
library(seqinr)
hoge <- read.fasta(in_f, seqtype="DNA")
hoge
hoge <- lapply(hoge, function(x){
translate(x, ambiguous=T)
})
hoge
write.fasta(hoge, names=names(hoge), file.out=out_f, nbchar=50)
3. (multi-)FASTA形式ファイル(sample4.fasta)の場合:
sapply関数を用いるやり方です。
in_f <- "sample4.fasta"
out_f <- "hoge3.fasta"
library(seqinr)
hoge <- read.fasta(in_f, seqtype="DNA")
hoge
hoge <- sapply(hoge, function(x){
translate(x, ambiguous=T)
})
hoge
write.fasta(hoge, names=names(hoge), file.out=out_f, nbchar=50)
4. (multi-)FASTA形式ファイル(sample4.fasta)の場合:
ファイルの入出力はBiostringsパッケージ、翻訳はseqinrパッケージを利用するやり方です。
Biostringとseqinrで同じtranslate関数が存在するため、
「seqinr::translate」として明示的にseqinrパッケージ中のtranslate関数を利用すると宣言しています。
in_f <- "sample4.fasta"
out_f <- "hoge4.fasta"
library(seqinr)
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge2 <- sapply(fasta, function(x){
hoge <- as.character(x)
c2s(seqinr::translate(s2c(hoge), ambiguous=T))
})
fasta <- AAStringSet(hoge2)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 相補鎖(complement)を取得
Biostringsパッケージ中のcomplement関数を用いて、FASTA形式ファイルを読み込んで相補鎖を得るやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample1.fasta"
out_f <- "hoge1.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- complement(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. multi-FASTA形式ファイル(h_rna.fasta)の場合:
in_f <- "h_rna.fasta"
out_f <- "hoge2.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- complement(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 逆相補鎖(reverse complement)を取得
Biostringsパッケージ中のreverseComplement関数を用いて、FASTA形式ファイルを読み込んで逆相補鎖を得るやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample1.fasta"
out_f <- "hoge1.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- reverseComplement(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. multi-FASTA形式ファイル(h_rna.fasta)の場合:
in_f <- "h_rna.fasta"
out_f <- "hoge2.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- reverseComplement(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 逆鎖(reverse)を取得
Biostringsパッケージ中のreverse関数を用いて、FASTA形式ファイルを読み込んで逆鎖を得るやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample1.fasta"
out_f <- "hoge1.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- reverse(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. multi-FASTA形式ファイル(h_rna.fasta)の場合:
in_f <- "h_rna.fasta"
out_f <- "hoge2.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- reverse(fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | k-mer解析 | k=1(塩基ごとの出現頻度解析) | Biostrings
Biostringsパッケージを用いて、multi-FASTA形式ファイルを読み込んで、"A", "C", "G", "T", ..., "N", ...など塩基ごとの出現頻度を調べるやり方を示します。
k-mer解析のk=1の場合に相当します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
配列ごとに出現頻度をカウントした結果を返すやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge1.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- alphabetFrequency(fasta)
out
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
1と基本的に同じで、出力結果を"A", "C", "G", "T", "N"のみに限定するやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge2.txt"
param_base <- c("A", "C", "G", "T", "N")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- alphabetFrequency(fasta)
obj <- is.element(colnames(hoge), param_base)
obj
out <- hoge[, obj]
out
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
配列ごとではなく、全配列をまとめて出現頻度をカウントした結果を返すやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge3.txt"
param_base <- c("A", "C", "G", "T", "N")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- alphabetFrequency(fasta)
obj <- is.element(colnames(hoge), param_base)
#out <- colSums(hoge[, obj])
out <- apply(as.matrix(hoge[, obj]), 2, sum)
tmp <- rbind(names(out), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
3と基本的に同じ結果ですが、転置させています。
in_f <- "hoge4.fa"
out_f <- "hoge4.txt"
param_base <- c("A", "C", "G", "T", "N")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- alphabetFrequency(fasta)
obj <- is.element(colnames(hoge), param_base)
#out <- colSums(hoge[, obj])
out <- apply(as.matrix(hoge[, obj]), 2, sum)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
multi-FASTAではないsingle-FASTA形式ファイルです。
colSums関数は行列データにしか適用できないが、apply(as.matrix...)とすることでmulti-FASTAでもsingle-FASTAでも統一的に取り扱えることがわかります。
と書いてますが挙動が変(R ver. 3.2.3)なので、変更予定です(2016年4月27日追加)。
in_f <- "sample1.fasta"
out_f <- "hoge5.txt"
param_base <- c("A", "C", "G", "T", "N")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- alphabetFrequency(fasta)
obj <- is.element(colnames(hoge), param_base)
out <- colSums(hoge[, obj])
#out <- apply(as.matrix(hoge[, obj]), 2, sum)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
multi-FASTAではないsingle-FASTA形式ファイルです。
colSums関数は行列データにしか適用できないが、apply(as.matrix...)とすることでmulti-FASTAでもsingle-FASTAでも統一的に取り扱えることがわかります。
と書いてますが挙動が変(R ver. 3.2.3)なので、変更予定です(2016年4月27日追加)。
in_f <- "sample1.fasta"
out_f <- "hoge6.txt"
param_base <- c("A", "C", "G", "T", "N")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- alphabetFrequency(fasta)
obj <- is.element(colnames(hoge), param_base)
#out <- colSums(hoge[, obj])
#out <- apply(as.matrix(hoge[, obj]), 2, sum)
out <- hoge[, obj]
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
DDBJ Pipeline (Nagasaki et al., DNA Res., 2013)上で
de novoゲノムアセンブリプログラムPlatanus (Kajitani et al., Genome Res., 2014)
を実行して得られたmulti-FASTA形式ファイル(out_gapClosed.fa; 約2.4MB)です。
in_f <- "out_gapClosed.fa"
out_f <- "hoge7.txt"
param_base <- c("A", "C", "G", "T", "N")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- alphabetFrequency(fasta)
obj <- is.element(colnames(hoge), param_base)
#out <- colSums(hoge[, obj])
out <- apply(as.matrix(hoge[, obj]), 2, sum)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
イントロ | 一般 | k-mer解析 | k=2(2連続塩基の出現頻度解析) | Biostrings
Biostringsパッケージを用いて、multi-FASTA形式ファイルを読み込んで、"AA", "AC", "AG", "AT", "CA", "CC", "CG", "CT", "GA", "GC", "GG", "GT", "TA", "TC", "TG", "TT"
の計42 = 16通りの2連続塩基の出現頻度を調べるやり方を示します。k-mer解析のk=2の場合に相当します。
ヒトゲノムで"CG"の割合が期待値よりも低い(Lander et al., 2001;
Saxonov et al., 2006)ですが、それを簡単に検証できます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
タイトル通りの出現頻度です。
in_f <- "hoge4.fa"
out_f <- "hoge1.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- dinucleotideFrequency(fasta)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
出現頻度ではなく、出現確率を得るやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge2.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3. multi-FASTA形式ファイル(h_rna.fasta)の場合:
406番目のID (NR_002762.1)の塩基配列の498番目の文字が"N"なはずなのですが、これはどのように計算されているのでしょうか。。。
in_f <- "h_rna.fasta"
out_f <- "hoge3.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- dinucleotideFrequency(fasta)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
タイトル通りの出現頻度です。イントロ | 一般 | 配列取得 | ゲノム配列 | BSgenome中のゲノム配列取得手順を含んでいます。
out_f <- "hoge4.txt"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
出現頻度ではなく、出現確率を得るやり方です。
out_f <- "hoge5.txt"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
出現頻度ではなく、出現確率を得るやり方です。パッケージがインストールされていない場合は、
インストール | Rパッケージ | 個別を参考にしてインストールしてから再度チャレンジ。
out_f <- "hoge6.txt"
param_bsgenome <- "BSgenome.Athaliana.TAIR.TAIR9"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2013年12月にリリースされたGenome Reference Consortium GRCh38です。出力は出現確率です。
out_f <- "hoge7.txt"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
全配列を合算して、連続塩基ごとの出現頻度(frequency)と出現確率(probability)を出力するやり方です。
dinucleotideFrequency関数中の「simplify.as="collapsed"」オプションでも一応実行できますが、
桁が多くなりすぎて「整数オーバーフロー」問題が起きたのでやめてます。
out_f <- "hoge8.txt"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
hoge <- dinucleotideFrequency(fasta, as.prob=F)
frequency <- colSums(hoge)
probability <- frequency / sum(frequency)
frequency
sort(frequency, decreasing=F)
tmp <- cbind(names(frequency), frequency, probability)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
基本的に7.と同じです。7.の手順がややこしいと思う人向けの解説用です。
簡単に言えば、パッケージ名を2回書かなくて済むテクニックを用いているだけです。
もう少し詳細に書くと、BSgenomeパッケージはlibrary関数で読み込んだ後にパッケージ名と同じ名前のオブジェクトを利用できるようになります。
例えばBSgenome.Hsapiens.NCBI.GRCh38パッケージの場合は、BSgenome.Hsapiens.NCBI.GRCh38
という名前のオブジェクトを利用できるようになります。
ベタで書くと2回BSgenome.Hsapiens.NCBI.GRCh38を記述する必要性があるため、間違う確率が上昇します。
7.のように一見ややこしく書けば、結果的に一度のみの記述で済むのです。
out_f <- "hoge9.txt"
library(Biostrings)
library(BSgenome.Hsapiens.NCBI.GRCh38)
tmp <- ls("package:BSgenome.Hsapiens.NCBI.GRCh38")
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
7.と基本的に同じですが、box plotのPNGファイルも出力しています。
out_f1 <- "hoge10.txt"
out_f2 <- "hoge10.png"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
param_fig <- c(700, 400)
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
boxplot(out, ylab="Probability")
grid(col="gray", lty="dotted")
dev.off()
10.と基本的に同じですが、連続塩基の種類ごとの期待値とボックスプロット(box plot)上での色情報を含むファイル
(human_2mer.txt)を入力として利用し、色情報のみを取り出して利用しています。
in_f <- "human_2mer.txt"
out_f1 <- "hoge11.txt"
out_f2 <- "hoge11.png"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
param_fig <- c(700, 400)
library(Biostrings)
library(param_bsgenome, character.only=T)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
boxplot(out, ylab="Probability", col=as.character(data$color))
grid(col="gray", lty="dotted")
dev.off()
11.と基本的に同じですが、human_2mer.txtというファイルを入力として与えて、
連続塩基の種類ごとの期待値とボックスプロット(box plot)上での色情報を利用しています。
また、重要なのは期待値からの差分であり、「プロットも期待値(expected)と同程度の観測値(observed)であればゼロ、
観測値のほうが大きければプラス、観測値のほうが小さければマイナス」といった具合で表現したほうがスマートです。
それゆえ、box plotの縦軸をlog(observed/expected)として表現しています。
CG以外の連続塩基は縦軸上でが0近辺に位置していることが分かります。
in_f <- "human_2mer.txt"
out_f1 <- "hoge12.txt"
out_f2 <- "hoge12.png"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
param_fig <- c(700, 400)
library(Biostrings)
library(param_bsgenome, character.only=T)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
out <- dinucleotideFrequency(fasta, as.prob=T)
logratio <- log2(out/data$expected)
tmp <- cbind(names(fasta), logratio)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
boxplot(logratio, ylab="log2(observed/expected)",
col=as.character(data$color))
grid(col="gray", lty="dotted")
dev.off()
イントロ | 一般 | k-mer解析 | k=3(3連続塩基の出現頻度解析) | Biostrings
Biostringsパッケージを用いて、43 = 64 通りの3連続塩基の出現頻度を調べるやり方を示します。k-mer解析のk=3の場合に相当します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "hoge4.fa"
out_f <- "hoge1.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- trinucleotideFrequency(fasta)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. 130MB程度のRefSeqのhuman mRNAのmulti-FASTAファイル(h_rna.fasta)の場合:
406番目のID (NR_002762.1)の塩基配列の498番目の文字が"N"なはずなのですが、これはどのように計算されているのでしょうか。。。
in_f <- "h_rna.fasta"
out_f <- "hoge2.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- trinucleotideFrequency(fasta)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
イントロ | 一般 | k-mer解析 | k=n(n連続塩基の出現頻度解析) | Biostrings
Biostringsパッケージを用いて、4n通りの任意のn連続塩基の出現頻度を調べるやり方を示します。k-mer解析のk=nの場合に相当します。
例えば2連続塩基の場合はk=2, 3連続塩基の場合はk=3と指定します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
4連続塩基(k=4)の出現頻度情報を得るやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge1.txt"
param_kmer <- 4
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- oligonucleotideFrequency(fasta, width=param_kmer)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2連続塩基(k=2)の出現頻度情報を得るやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge2.txt"
param_kmer <- 2
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- oligonucleotideFrequency(fasta, width=param_kmer)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2連続塩基(k=2)の全配列をまとめた出現頻度情報を得るやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge3.txt"
param_kmer <- 2
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- oligonucleotideFrequency(fasta, width=param_kmer, simplify.as="collapsed")
tmp <- cbind(names(out), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2連続塩基(k=2)の全配列をまとめた出現確率情報を得るやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge4.txt"
param_kmer <- 2
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- oligonucleotideFrequency(fasta, width=param_kmer, simplify.as="collapsed", as.prob=TRUE)
tmp <- cbind(names(out), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
10連続塩基(k=10)の出現頻度情報を得るやり方です。4^10 = 1,048,576(105万)通りのk-merの出現頻度を計算することになります。
in_f <- "hoge4.fa"
out_f <- "hoge5.txt"
param_kmer <- 10
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- oligonucleotideFrequency(fasta, width=param_kmer)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
15連続塩基(k=15)の出現頻度情報を得るやり方です。4^15 = 1,073,741,824(約11億)通りのk-merの出現頻度を計算することになります。
8GBメモリマシンで実行すると、「エラー: サイズ 16.0 Gb のベクトルを割り当てることができません」となります。
in_f <- "hoge4.fa"
out_f <- "hoge6.txt"
param_kmer <- 15
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- oligonucleotideFrequency(fasta, width=param_kmer)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2連続塩基(k=2)の出現頻度情報を得るやり方です。4^2 = 16通りのk-merの出現頻度を計算することになります。
リード毎に出現頻度を算出しています。
in_f <- "sample32_ngs.fasta"
out_f <- "hoge7.txt"
param_kmer <- 2
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out <- oligonucleotideFrequency(fasta, width=param_kmer)
tmp <- cbind(names(fasta), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2連続塩基(k=2)の出現頻度情報を得るやり方です。4^2 = 16通りのk-merの出現頻度を計算することになります。
全リードを合算した出現頻度を出力するやり方です。
in_f <- "sample32_ngs.fasta"
out_f <- "hoge8.txt"
param_kmer <- 2
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- oligonucleotideFrequency(fasta, width=param_kmer)
out <- colSums(hoge)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
3連続塩基(k=3)の出現頻度情報を得るやり方です。4^3 = 64通りのk-merの出現頻度を計算することになります。
全リードを合算した出現頻度を出力するやり方です。
in_f <- "sample32_ngs.fasta"
out_f <- "hoge9.txt"
param_kmer <- 3
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- oligonucleotideFrequency(fasta, width=param_kmer)
out <- colSums(hoge)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
3連続塩基(k=3)の出現頻度情報を得るやり方です。4^3 = 64通りのk-merの出現頻度を計算することになります。
全リードを合算した出現頻度を出力するやり方です。
in_f <- "sample33_ngs.fasta"
out_f <- "hoge10.txt"
param_kmer <- 3
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- oligonucleotideFrequency(fasta, width=param_kmer)
out <- colSums(hoge)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
10連続塩基(k=10)の出現頻度情報を得るやり方です。4^10 = 1,048,576通りのk-merの出現頻度を計算することになります。
全リードを合算した出現頻度を出力するやり方です。
in_f <- "sample33_ngs.fasta"
out_f <- "hoge11.txt"
param_kmer <- 10
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- oligonucleotideFrequency(fasta, width=param_kmer)
out <- colSums(hoge)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
length(out)
sum(out > 0)
10連続塩基(k=10)の出現頻度情報を得るやり方です。4^10 = 1,048,576通りのk-merの出現頻度を計算することになります。
全リードを合算した出現頻度を出力するやり方です。
in_f <- "sample34_ngs.fasta"
out_f <- "hoge12.txt"
param_kmer <- 10
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- oligonucleotideFrequency(fasta, width=param_kmer)
out <- colSums(hoge)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
length(out)
sum(out > 0)
イントロ | 一般 | Tips | 任意の拡張子名でファイルを保存
出力ファイル名をわざわざ指定せずに、拡張子名のみ変えるやり方を示します。。(例:XXX.txtを読み込んでXXX.inuという拡張子で保存したい)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
*.ugeというファイル名で保存するやり方です。
in_f <- "hoge4.fa"
param <- "uge"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
out_f <- paste(unlist(strsplit(in_f, ".", fixed=TRUE))[1], param, sep=".")
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | Tips | 拡張子は同じで任意の文字を追加して保存
ファイル保存時に、拡張子名は変えずに、拡張子の手前に任意の文字(例:"_processed")を追加するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
*_processed.faというファイル名で保存するやり方です。
ファイル名中に"."(ドット)が一つしかないという前提です
in_f <- "hoge4.fa"
param <- "_processed"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge1 <- paste(".", unlist(strsplit(in_f, ".", fixed=TRUE))[2], sep="")
hoge2 <- paste(param, hoge1, sep="")
out_f <- sub(hoge1, hoge2, in_f)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 配列取得 | ゲノム配列 | 公共DBから
イントロ | 一般 | 配列取得 | ゲノム配列 | BSgenome
BSgenomeパッケージを用いて様々な生物種のゲノム配列を取得するやり方を示します。
ミヤマハタザオ (A. lyrata)、セイヨウミツバチ (A. mellifera)、
シロイヌナズナ(A.thaliana)、ウシ(B.taurus)、線虫(C.elegans)、犬(C.familiaris)、キイロショウジョウバエ(D.melanogaster)、
ゼブラフィッシュ(D.rerio)、大腸菌(E.coli)、イトヨ(G.aculeatus)、セキショクヤケイ(G.gallus)、ヒト(H.sapiens)、
アカゲザル(M.mulatta)、マウス(M.musculus)、チンパンジー(P.troglodytes)、ラット(R.norvegicus)、出芽酵母(S.cerevisiae)、
トキソプラズマ(T.gondii)と実に様々な生物種が利用可能であることがわかります。
getSeq関数はBSgenomeオブジェクト中の「single sequences」というあたりにリストアップされているchr...というものを全て抽出しています。
したがって、例えばマウスゲノムは「chr1」以外に「chr1_random」や「chrUn_random」なども等価に取扱っている点に注意してください。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 利用可能な生物種とRにインストール済みの生物種をリストアップしたい場合:
library(BSgenome)
available.genomes()
installed.genomes()
installed.genomes(splitNameParts=TRUE)
2. ゼブラフィッシュ("BSgenome.Drerio.UCSC.danRer7")のゲノム情報をRにインストールしたい場合:
400MB程度あります...
param <- "BSgenome.Drerio.UCSC.danRer7"
source("http://bioconductor.org/biocLite.R")
biocLite(param)
installed.genomes()
3. インストール済みのゼブラフィッシュのゲノム配列をmulti-FASTAファイルで保存したい場合:
1.4GB程度のファイルが生成されます...
out_f <- "hoge3.fasta"
param <- "BSgenome.Drerio.UCSC.danRer7"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. インストール済みのマウス("BSgenome.Mmusculus.UCSC.mm9")のゲノム配列をmulti-FASTAファイルで保存したい場合:
2.8GB程度のファイルが生成されます...
out_f <- "hoge4.fasta"
param <- "BSgenome.Mmusculus.UCSC.mm9"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. インストール済みのヒト("BSgenome.Hsapiens.UCSC.hg19")のゲノム配列をmulti-FASTAファイルで保存したい場合:
3.0GB程度のファイルが生成されます...。ヒトゲノムは、まだ完全に22本の常染色体とX, Y染色体の計24本になっているわけではないことがわかります。
out_f <- "hoge5.fasta"
param <- "BSgenome.Hsapiens.UCSC.hg19"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
6. インストール済みのヒト("BSgenome.Hsapiens.UCSC.hg19")のゲノム配列をmulti-FASTAファイルで保存したい場合:
ヒトゲノムは、まだ完全に22本の常染色体とX, Y染色体の計24本になっているわけではないので、最初の主要な24本分のみにするやり方です。
out_f <- "hoge6.fasta"
param1 <- "BSgenome.Hsapiens.UCSC.hg19"
param2 <- 24
library(param1, character.only=T)
#tmp <- unlist(strsplit(param1, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param1, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta <- fasta[1:param2]
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
8. インストール済みのシロイヌナズナ("BSgenome.Athaliana.TAIR.TAIR9")のゲノム配列をmulti-FASTAファイルで保存したい場合:
The Arabidopsis Information Resource (TAIR)(Reiser et al., Curr Protoc Bioinformatics., 2017)
から得られる最新バージョンはTAIR10ですが、アセンブリ結果自体はTAIR9と同じと明記されています
(README_whole_chromosomes.txt)。
120MB程度のファイルが生成されます...
out_f <- "hoge8.fasta"
param <- "BSgenome.Athaliana.TAIR.TAIR9"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
9. インストール済みのヒト("BSgenome.Hsapiens.NCBI.GRCh38")のゲノム配列をmulti-FASTAファイルで保存したい場合:
2013年12月にリリースされたGenome Reference Consortium GRCh38です。
R ver. 3.1.0とBioconductor ver. 2.14以上の環境で実行可能です。
out_f <- "hoge9.fasta"
param <- "BSgenome.Hsapiens.NCBI.GRCh38"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
10. インストール済みのヒト("BSgenome.Hsapiens.NCBI.GRCh38")のゲノム配列のmulti-FASTAファイルで保存したい場合:
一部を抽出して保存するやり方です。このパッケージ中の染色体の並びが既知(chr1, 2, ..., chr22, chrX, chrY, and MT)であるという前提です。
out_f <- "hoge10.fasta"
param <- "BSgenome.Hsapiens.NCBI.GRCh38"
param_range <- 1:25
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
obj <- param_range
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
11. インストール済みのヒト("BSgenome.Hsapiens.UCSC.hg38")のゲノム配列をmulti-FASTAファイルで保存したい場合:
3.0GB程度のファイルが生成されます...。ヒトゲノムは、まだ完全に22本の常染色体とX, Y染色体の計24本になっているわけではないことがわかります。
out_f <- "hoge11.fasta"
param <- "BSgenome.Hsapiens.UCSC.hg38"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
genome
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
ゲノム配列(BSgenome)パッケージとアノテーション情報(TxDb)パッケージ
を用いて様々な生物種のプロモーター配列(転写開始点近傍配列;上流配列)を取得するやり方を示します。
2014年4月リリースのBioconductor 2.14以降の推奨手順では、ゲノムのパッケージ(例:BSgenome.Hsapiens.UCSC.hg19)と
対応するアノテーションパッケージ(例:TxDb.Hsapiens.UCSC.hg19.knownGene)の両方を読み込ませる必要がありますので、2015年2月に記述内容を大幅に変更しました。
ヒトなどの主要なパッケージ以外はおそらくデフォルトではインストールされていませんので、「パッケージがインストールされていません」的なエラーが出た場合は、
個別パッケージのインストールを参考にして予め利用したいパッケージのインストールを行ってから再挑戦してください。
出力はmulti-FASTAファイルです。
現状では、ゼブラフィッシュ(danRer7)はゲノムパッケージ(BSgenome.Drerio.UCSC.danRer7)
は存在しますが、対応するTxDbパッケージが存在しないので、どこかからGFFファイルを取得してmakeTxDbFromGFF関数などを利用してTxDbオブジェクトを得るなどする必要があります。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. ヒト(hg19)の場合:
ゲノムパッケージ(BSgenome.Hsapiens.UCSC.hg19)と
対応するアノテーションパッケージ(TxDb.Hsapiens.UCSC.hg19.knownGene)
を読み込んで、転写開始点上流1000塩基分を取得するやり方です。
out_f <- "hoge1.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
param_upstream <- 1000
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
gn <- sort(genes(txdb))
hoge <- flank(gn, width=param_upstream)
fasta <- getSeq(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. ヒト(hg19)の場合:
ゲノムパッケージ(BSgenome.Hsapiens.UCSC.hg19)と
対応するアノテーションパッケージ(TxDb.Hsapiens.UCSC.hg19.knownGene)
を読み込んで、転写開始点上流500塩基から下流20塩基までの範囲を取得するやり方です。
out_f <- "hoge2.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
param_upstream <- 500
param_downstream <- 20
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
gn <- sort(genes(txdb))
hoge <- promoters(gn, upstream=param_upstream,
downstream=param_downstream)
fasta <- getSeq(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. 線虫(ce6)の場合:
ゲノムパッケージ(BSgenome.Celegans.UCSC.ce6)と
対応するアノテーションパッケージ(TxDb.Celegans.UCSC.ce6.ensGene)
を読み込んで、転写開始点上流500塩基から下流20塩基までの範囲を取得するやり方です。
out_f <- "hoge3.fasta"
param_bsgenome <- "BSgenome.Celegans.UCSC.ce6"
param_txdb <- "TxDb.Celegans.UCSC.ce6.ensGene"
param_upstream <- 500
param_downstream <- 20
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
gn <- sort(genes(txdb))
hoge <- promoters(gn, upstream=param_upstream,
downstream=param_downstream)
fasta <- getSeq(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. マウス(mm10)の場合:
ゲノムパッケージ(BSgenome.Mmusculus.UCSC.mm10)と
対応するアノテーションパッケージ(TxDb.Mmusculus.UCSC.mm10.knownGene)
を読み込んで、転写開始点上流500塩基から下流20塩基までの範囲を取得するやり方です。
out_f <- "hoge4.fasta"
param_bsgenome <- "BSgenome.Mmusculus.UCSC.mm10"
param_txdb <- "TxDb.Mmusculus.UCSC.mm10.knownGene"
param_upstream <- 500
param_downstream <- 20
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
gn <- sort(genes(txdb))
hoge <- promoters(gn, upstream=param_upstream,
downstream=param_downstream)
fasta <- getSeq(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. ヒト(hg38)の場合:
ゲノムパッケージ(BSgenome.Hsapiens.NCBI.GRCh38)と
対応するアノテーションパッケージ(TxDb.Hsapiens.UCSC.hg38.knownGene)
を読み込んで、転写開始点上流500塩基から下流20塩基までの範囲を取得するやり方として書いてはいますが、
「sequence chr1 not found」のようなエラーが出ます。この理由は、情報提供元が異なるため同一染色体の対応付けができないことに起因します。
out_f <- "hoge5.fasta"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
param_txdb <- "TxDb.Hsapiens.UCSC.hg38.knownGene"
param_upstream <- 500
param_downstream <- 20
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
gn <- sort(genes(txdb))
hoge <- promoters(gn, upstream=param_upstream,
downstream=param_downstream)
fasta <- getSeq(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
GenomicFeaturesパッケージを主に用いてプロモーター配列(転写開始点近傍配列)を得るやり方を示します。
ここでは、イントロ | 一般 | 配列取得 | ゲノム配列(BSgenomeから)で指定可能なゲノムと
イントロ | NGS | アノテーション情報取得 | TxDb | GenomicFeatures(Lawrence_2013)で作成可能なTxDbオブジェクトを入力として、
転写開始点から任意の[上流xxx塩基, 下流yyy塩基]分の塩基配列を取得して、
FASTA形式ファイルで保存するやり方を示しています。makeTxDbFromGFF関数は、どの生物種に対してもデフォルトではChrMのみ環状ゲノムとするようですが、
circ_seqsオプションで任意の染色体の環状・非環状の指定が可能です(高橋 広夫 氏提供情報)。
ちなみに、ゲノムの方の環状・非環状の指定のやり方は、例えば「Chr1, Chr2, Chr3, Chr4, Chr5, ChrM, ChrC」のシロイヌナズナゲノムのオブジェクトgenomeに対して
ChrMのみ環状としたい場合は、genome@seqinfo@is_circular <- c(F, F, F, F, F, T, F)のように指定すればよいです。
multi-FASTA形式のゲノム配列ファイルとGFF3形式のアノテーションファイルのみからプロモータ配列を取得するやり方も示しています。
2017年6月23日に、例題12のコードを、コンティグ(配列)数が複数で「FASTAファイルには存在するがGFFファイル中には存在しない配列があった場合」に不都合が生じる問題を回避できるように書き換えました(野間口達洋氏 提供情報)。
1. ヒト("BSgenome.Hsapiens.UCSC.hg19")の[上流200塩基, 下流30塩基]のプロモーター配列を取得する場合:
ヒトゲノム ver. 19 ("hg19"; Genome Reference Consortium GRCh37のことらしい)のUCSC Genes ("knownGene")のTxDbオブジェクトを利用しています。
out_f <- "hoge1.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb1 <- "hg19"
param_txdb2 <- "knownGene"
param_upstream <- 200
param_downstream <- 30
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
txdb <- makeTxDbFromUCSC(genome=param_txdb1, tablename=param_txdb2)
txdb
out <- getPromoterSeq(transcriptsBy(txdb, by="gene"),
genome, upstream=param_upstream, downstream=param_downstream)
out
fasta <- unlist(out)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. ヒト("BSgenome.Hsapiens.UCSC.hg19")の[上流200塩基, 下流30塩基]のプロモーター配列を取得する場合:
ヒトゲノム ver. 19 ("hg19"; Genome Reference Consortium GRCh37のことらしい)のEnsembl Genes ("ensGene")のTxDbオブジェクトを利用しています。
out_f <- "hoge2.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb1 <- "hg19"
param_txdb2 <- "ensGene"
param_upstream <- 200
param_downstream <- 30
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
txdb <- makeTxDbFromUCSC(genome=param_txdb1, tablename=param_txdb2)
txdb
out <- getPromoterSeq(transcriptsBy(txdb, by="gene"),
genome, upstream=param_upstream, downstream=param_downstream)
out
fasta <- unlist(out)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. シロイヌナズナ("BSgenome.Athaliana.TAIR.TAIR9")の[上流500塩基, 下流0塩基]のプロモーター配列を取得する場合:
UCSCからはArabidopsisの遺伝子アノテーション情報が提供されていないため、
TAIR10_GFF3_genes.gffを予めダウンロードしておき、
makeTxDbFromGFF関数を用いてTxDbオブジェクトを作成しています。
この関数は、どの生物種に対してもデフォルトではChrMのみ環状ゲノムとするようですので、
circ_seqsオプションで葉緑体ゲノム(Chloroplast; ChrC)が環状であると認識させるやり方です(高橋 広夫 氏提供情報)。
これでゲノムの環状・非環状に起因するエラーを解決できたので第一段階突破です(2015.03.04追加)。
それでも「以下にエラー loadFUN(x, seqname, ranges) : trying to load regions beyond the boundaries of non-circular sequence "Chr3"」
というエラーに遭遇してしまいます。この解決策は4.に記載しています。(2015.08.03追加)
in_f <- "TAIR10_GFF3_genes.gff"
out_f <- "hoge3.fasta"
param_bsgenome <- "BSgenome.Athaliana.TAIR.TAIR9"
param_upstream <- 500
param_downstream <- 0
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
seqinfo(genome)
txdb <- makeTxDbFromGFF(in_f, format="gff3", circ_seqs=c("ChrC","ChrM"))
txdb
seqinfo(txdb)
out <- getPromoterSeq(transcriptsBy(txdb, by="gene"),
genome, upstream=param_upstream, downstream=param_downstream)
out
fasta <- unlist(out)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. シロイヌナズナ("BSgenome.Athaliana.TAIR.TAIR9")の[上流500塩基, 下流0塩基]のプロモーター配列を取得する場合:
3.のエラーへの対処法(甲斐政親氏 提供情報)。取得予定の範囲が完全にゲノム配列内に収まっているもののみ抽出してから
getSeq関数を用いて配列取得しています。(2015.08.03追加)
in_f <- "TAIR10_GFF3_genes.gff"
out_f <- "hoge4.fasta"
param_bsgenome <- "BSgenome.Athaliana.TAIR.TAIR9"
param_upstream <- 500
param_downstream <- 0
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
seqinfo(genome)
txdb <- makeTxDbFromGFF(in_f, format="gff3", circ_seqs=c("ChrC","ChrM"))
txdb
seqinfo(txdb)
#gn <- sort(genes(txdb))
hoge <- promoters(txdb, upstream=param_upstream,
downstream=param_downstream)
hoge
hoge3 <- param_upstream + param_downstream
obj <- sapply(hoge,
function(hoge2) {
start <- hoge2@ranges[[1]][1]
end <- hoge2@ranges[[1]][hoge3]
chr_num <- grep(paste(hoge2@seqnames@values, "$", sep=""), hoge@seqnames@values)
chr_length <- genome[[chr_num]]@length
if ((start >= 0) & (end <= chr_length)){
return(TRUE)
} else {
return(FALSE)
}
})
hoge <- hoge[obj]
hoge
fasta <- getSeq(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. シロイヌナズナ(TAIR10_chr_all.fas.gz)
の[上流500塩基, 下流0塩基]のプロモーター配列を取得する場合:
UCSCからはArabidopsisの遺伝子アノテーション情報が提供されていないため、
TAIR10_GFF3_genes.gffを予めダウンロードしておき、
makeTxDbFromGFF関数を用いてTxDbオブジェクトを作成しています。
イントロ | NGS | 読み込み | FASTA形式 | description行の記述を整形を参考にして、
description行の文字列をgffファイルと対応がとれるように変更しています。
2023年4月に再度実行させると不具合が生じるようになっていることがわかりましたので、「前処理(欲しい領域がゲノム配列の範囲内のもののみ抽出)」の部分を修正しています(2023.04.21追加)。
in_f1 <- "TAIR10_chr_all.fas.gz"
in_f2 <- "TAIR10_GFF3_genes.gff"
out_f <- "hoge5.fasta"
param_upstream <- 500
param_downstream <- 0
library(Rsamtools)
library(Biostrings)
library(GenomicFeatures)
fasta <- readDNAStringSet(in_f1, format="fasta")
fasta
uge <- strsplit(x = names(fasta), split = " ")
names(fasta) <- sapply(uge, `[`, 1)
fasta
writeXStringSet(fasta, file="hogegege.fa", format="fasta", width=50)
txdb <- makeTxDbFromGFF(in_f2, format="gff3", circ_seqs=c("ChrC","ChrM"))
txdb
hoge <- promoters(txdb, upstream=param_upstream,
downstream=param_downstream)
hoge
start <- hoge@ranges@start
end <- start + hoge@ranges@width - 1
chrlen <- unlist(mapply(rep, width(fasta), hoge@seqnames@lengths))
obj <- (start >= 0) & (end <= chrlen)
hoge <- hoge[obj]
hoge
fasta <- getSeq(FaFile("hogegege.fa"), hoge)
fasta
names(fasta) <- names(hoge)
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
6. ヒト("BSgenome.Hsapiens.NCBI.GRCh38")の[上流200塩基, 下流30塩基]のプロモーター配列を取得する場合:
ヒトゲノム ("hg38"; 2013年12月にリリースされたGenome Reference Consortium GRCh38)です。
現状では、RefSeq Genes ("refGene")を指定するとTxDbオブジェクト作成まではできますがgetPromoterSeq関数実行時にエラーが出ます。
また、Ensembl Genes ("ensGene")を指定するとTxDbオブジェクト作成段階でエラーが出ます...orz
out_f <- "hoge6.fasta"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
param2 <- "hg38"
param3 <- "refGene"
param_upstream <- 200
param_downstream <- 30
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
txdb <- makeTxDbFromUCSC(genome=param2, tablename=param3)
txdb
out <- getPromoterSeq(transcriptsBy(txdb, by="gene"),
genome, upstream=param_upstream, downstream=param_downstream)
out
fasta <- unlist(out)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)と
GFF3形式のアノテーションファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3)
を読み込むやり方です。「前処理(欲しい領域の座標情報取得)」の部分の条件判定は、start部分のみで中途半端ですのでご注意ください(2023.04.21追加)。
in_f1 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f <- "hoge7.fasta"
param_upstream <- 500
param_downstream <- 20
library(Rsamtools)
library(Biostrings)
library(GenomicFeatures)
#txdb <- makeTxDbFromGFF(in_f2,
# format="gff3", useGenesAsTranscripts=T)
txdb <- makeTxDbFromGFF(in_f2, format="gff3")
txdb
gn <- sort(genes(txdb))
hoge <- promoters(gn, upstream=param_upstream,
downstream=param_downstream)
hoge
obj <- (ranges(hoge)@start >= 0)
hoge <- hoge[obj]
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
names(fasta) <- names(hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
7.と基本的に同じですが、少なくとも2015年4月リリースのBioconductor ver. 3.1以降で、
GenomicFeatures
パッケージが提供するpromoters関数がTxDbオブジェクトをそのまま読み込めることが判明したので、それを反映させています。
in_f1 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f <- "hoge8.fasta"
param_upstream <- 100
param_downstream <- 0
library(Rsamtools)
library(Biostrings)
library(GenomicFeatures)
#txdb <- makeTranscriptDbFromGFF(in_f2,
# format="gff3", useGenesAsTranscripts=T)
txdb <- makeTxDbFromGFF(in_f2, format="gff3")
txdb
#gn <- sort(genes(txdb))
hoge <- promoters(txdb, upstream=param_upstream,
downstream=param_downstream)
hoge
obj <- (ranges(hoge)@start >= 0)
hoge <- hoge[obj]
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
names(fasta) <- names(hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
8.と基本的に同じですが、環状ゲノムの場合はmakeTxDbFromGFF関数実行時にcirc_seqsオプションで該当染色体(or 配列)を指定する必要があります。
ここに記載する文字列は、基本的にゲノムファイルのdescription行部分に記載されているものと同じ(つまり乳酸菌の場合は"Chromosome")である必要があります。
そのため灰色で「前処理(ゲノムファイルのdescription行情報を把握)」
in_f1 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f <- "hoge9.fasta"
param_upstream <- 100
param_downstream <- 0
library(Rsamtools)
library(Biostrings)
library(GenomicFeatures)
fasta <- readDNAStringSet(in_f1, format="fasta")
fasta
txdb <- makeTxDbFromGFF(in_f2, format="gff3", circ_seqs="Chromosome")
txdb
hoge <- promoters(txdb, upstream=param_upstream,
downstream=param_downstream)
hoge
obj <- (ranges(hoge)@start >= 0)
hoge <- hoge[obj]
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
names(fasta) <- names(hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
10. GFF3形式のアノテーションファイルとFASTA形式のゲノム配列ファイルを読み込む場合:
GFF3形式ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3)
とFASTA形式ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa)を読み込むやり方です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus hokkaidonensis JCM 18461 (Tanizawa et al., 2015)
のデータです。
in_f1 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3"
out_f <- "hoge10.fasta"
param_upstream <- 100
param_downstream <- 10
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")
txdb
hoge <- promoters(txdb, upstream=param_upstream,
downstream=param_downstream)
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
fasta
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
11. GFF3形式のアノテーションファイルとFASTA形式のゲノム配列ファイルを読み込む場合:
10.と基本的に同じでparam_upstreamのところを200に変更しているだけですが、エラーが出ることがわかります。
理由は、存在しないゲノム領域からプロモーター配列を取得しようとしたからです。
in_f1 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3"
out_f <- "hoge11.fasta"
param_upstream <- 200
param_downstream <- 10
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")
txdb
hoge <- promoters(txdb, upstream=param_upstream,
downstream=param_downstream)
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
fasta
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
12. GFF3形式のアノテーションファイルとFASTA形式のゲノム配列ファイルを読み込む場合:
11.と基本的に同じですが、取得予定領域座標がゲノム配列の範囲外にあるものをフィルタリングする部分を追加しています(甲斐政親氏 提供情報)。
この例題コードだと問題ないのですが、コンティグ(配列)数が複数で「FASTAファイルには存在するがGFFファイル中には存在しない配列があった場合」に不都合が生じるので、2017年6月23日に
"hoge@seqnames@values"だったところを"names(fasta)"に変更しました(野間口達洋 氏提供情報)。
in_f1 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3"
out_f <- "hoge12.fasta"
param_upstream <- 200
param_downstream <- 10
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto", circ_seqs=c("Chromosome"))
txdb
fasta <- readDNAStringSet(in_f1, format="fasta")
hoge <- promoters(txdb, upstream=param_upstream,
downstream=param_downstream)
hoge
hoge3 <- param_upstream + param_downstream
obj <- sapply(hoge,
function(hoge2) {
start <- hoge2@ranges[[1]][1]
end <- hoge2@ranges[[1]][hoge3]
chr_num <- grep(paste(hoge2@seqnames@values, "$", sep=""), names(fasta))
chr_length <- seqlengths(FaFile(in_f1))[chr_num]
if ((start >= 0) & (end <= chr_length)){
return(TRUE)
} else {
return(FALSE)
}
})
hoge <- hoge[obj]
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
fasta
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 配列取得 | トランスクリプトーム配列 | GenomicFeatures(Lawrence_2013)
GenomicFeaturesパッケージを主に用いてトランスクリプトーム配列を得るやり方を示します。
「?extractTranscriptSeqs」を行うことで、様々な例題を見ることができます。transcriptsBy関数部分は、exonsBy, cdsBy, intronsByTranscript, fiveUTRsByTranscript, threeUTRsByTranscript
など様々な他の関数で置き換えることができます。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
対応するアノテーションパッケージ(TxDb.Hsapiens.UCSC.hg19.knownGene)を利用しています。基本形です。
2015年3月2日の実行結果は82,960 transcriptsでした。
out_f <- "hoge1.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
txdb
fasta <- extractTranscriptSeqs(genome, txdb)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
extractTranscriptSeqsに与えている情報の形式がtxdbからhogeオブジェクトに変わっているだけです。
2015年3月2日現在、transcriptsBy関数実行段階で「いくつかの転写物が複数の染色体上にまたがるexonを持つ。
現状ではこのようなものに対応していない。」的なエラーメッセージが出ます。
out_f <- "hoge2.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
txdb
hoge <- transcriptsBy(txdb, by="gene")
fasta <- extractTranscriptSeqs(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2.とほぼ同じですが、transcriptsBy関数実行時のオプションとしてexonを与えています。
非常に計算が大変らしく、メモリ8GBのノートPCではメモリが足りないと文句を言われて実行できません(爆)。
out_f <- "hoge3.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
txdb
hoge <- transcriptsBy(txdb, by="exon")
fasta <- extractTranscriptSeqs(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
cdsBy関数をby="gene"オプションで実行するやり方ですが、2.の結果と同じくエラーが出ます。
out_f <- "hoge4.fasta"
param_bsgenome <- "BSgenome.Hsapiens.UCSC.hg19"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
library(GenomicFeatures)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
genome
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
txdb
hoge <- cdsBy(txdb, by="gene")
fasta <- extractTranscriptSeqs(genome, hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. GFF3形式のアノテーションファイルとFASTA形式のゲノム配列ファイルを読み込む場合:
GFF3形式ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3)
とFASTA形式ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa)を読み込むやり方です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus hokkaidonensis JCM 18461 (Tanizawa et al., 2015)
のデータです。
in_f1 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3"
out_f <- "hoge5.fasta"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")
txdb
hoge <- transcripts(txdb)
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
fasta
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
6. GFF3形式のアノテーションファイルとFASTA形式のゲノム配列ファイルを読み込む場合:
GFF3形式ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3)
とFASTA形式ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)を読み込むやり方です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aです。
in_f1 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f <- "hoge6.fasta"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")
txdb
hoge <- transcripts(txdb)
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
fasta
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 配列取得 | トランスクリプトーム配列 | biomaRt(Durinck_2009)
biomaRtパッケージ
(Durinck et al., Nat Protoc., 2009)を用いて様々なIDに対応する転写物(トランスクリプトーム)配列を取得するやり方を示します。
BioMart (Kasprzyk A., Database, 2011)本体のウェブサービスへのアクセスをR経由で行うもの、という理解でいいです。2022.04.26に多少修正しましたが、まだうまく取得できないですのでご注意ください。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. RefSeq mRNAをキーとしてヒトのcDNA配列を取得したい場合:
out_f <- "hoge1.fasta"
param_dataset <- "hsapiens_gene_ensembl"
param_attribute <- "refseq_mrna"
param_seqtype <- "cdna"
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
hoge <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(hoge)
dim(hoge)
out <- getSequence(id=hoge[,1], type=param_attribute,
seqType=param_seqtype, mart=mart)
dim(out)
exportFASTA(out, file=out_f)
2. Ensembl Gene IDをキーとしてヒトのcDNA配列を取得したい場合:
out_f <- "hoge2.fasta"
param_dataset <- "hsapiens_gene_ensembl"
param_attribute <- "ensembl_gene_id"
param_seqtype <- "cdna"
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
hoge <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(hoge)
dim(hoge)
out <- getSequence(id=hoge[,1], type=param_attribute,
seqType=param_seqtype, mart=mart)
dim(out)
exportFASTA(out, file=out_f)
3. Ensembl Gene IDをキーとしてヒトのgene_exon_intron配列を取得したい場合:
out_f <- "hoge3.fasta"
param_dataset <- "hsapiens_gene_ensembl"
param_attribute <- "ensembl_gene_id"
param_seqtype <- "gene_exon_intron"
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
hoge <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(hoge)
dim(hoge)
out <- getSequence(id=hoge[,1], type=param_attribute,
seqType=param_seqtype, mart=mart)
dim(out)
exportFASTA(out, file=out_f)
4. RefSeq mRNAをキーとしてラットのcDNA配列を取得したい場合:
out_f <- "hoge4.fasta"
param_dataset <- "rnorvegicus_gene_ensembl"
param_attribute <- "refseq_mrna"
param_seqtype <- "cdna"
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
hoge <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(hoge)
dim(hoge)
out <- getSequence(id=hoge[,1], type=param_attribute,
seqType=param_seqtype, mart=mart)
dim(out)
exportFASTA(out, file=out_f)
5. EMBL (Genbank) IDをキーとしてモルモット(C. porcellus)の5' UTR配列を取得したい場合:
out_f <- "hoge5.fasta"
param_dataset <- "cporcellus_gene_ensembl"
param_attribute <- "embl"
param_seqtype <- "5utr"
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
hoge <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(hoge)
dim(hoge)
out <- getSequence(id=hoge[,1], type=param_attribute,
seqType=param_seqtype, mart=mart)
dim(out)
exportFASTA(out, file=out_f)
イントロ | 一般 | 配列取得 | CDS | GenomicFeatures(Lawrence_2013)
GenomicFeaturesパッケージを主に用いてCDS(タンパク質コード領域の配列)を得るやり方を示します。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. GFF3形式のアノテーションファイルとFASTA形式のゲノム配列ファイルを読み込む場合:
GFF3形式ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3)
とFASTA形式ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)を読み込むやり方です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aです。2,681個の配列が取得できます。
in_f1 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f <- "hoge1.fasta"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")
txdb
hoge <- cds(txdb)
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
fasta
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. GFF3形式のアノテーションファイルとFASTA形式のゲノム配列ファイルを読み込む場合:
2019年5月13日の講義で利用した、GFF3形式ファイル(annotation.gff)
とFASTA形式ファイル(genome.fna)を読み込むやり方です。
エラーは出ていないものの、GFFファイルをうまく読み込めていないようです。
つまり、txdbのところです。gffをgff3に変えるとか、fnaをfaにするとかそういう問題ではなさそうです。
正解は、(description行部分が異なりますが)cds.fnaです。
in_f1 <- "genome.fna"
in_f2 <- "annotation.gff"
out_f <- "hoge2.fasta"
library(Rsamtools)
library(GenomicFeatures)
library(Biostrings)
txdb <- makeTxDbFromGFF(in_f2, format="auto")
txdb
hoge <- cds(txdb)
hoge
fasta <- getSeq(FaFile(in_f1), hoge)
fasta
names(fasta) <- paste(seqnames(hoge), start(ranges(hoge)),
end(ranges(hoge)), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | 一般 | 読み込み | xlsx形式 | openxlsx
openxlsxパッケージを用いて、Microsoft EXCEL (マイクロソフトエクセル)の
.xlsxという拡張子のついたファイルを読み込むやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.xlsx"
out_f <- "hoge1.txt"
library(openxlsx)
data <- read.xlsx(in_f, colNames=T, rowNames=T)
head(data)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
1.と基本的に同じですが、余分な情報を最初の2行分に含みます。このため、最初の3行目から読み込むやり方です。
in_f <- "data_hypodata_3vs3_plus.xlsx"
out_f <- "hoge2.txt"
param_skip <- 3
library(openxlsx)
data <- read.xlsx(in_f, startRow=param_skip, colNames=T, rowNames=T)
head(data)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Additional file 1として提供されているxlsxファイルです。
2群間比較用(8 samples vs. 8 samples)データです。余分な情報を最初の1行分に含みます。
このため、最初の2行目から読み込むやり方です。
#in_f <- "http://www.biomedcentral.com/content/supplementary/s12864-015-1767-y-s1.xlsx"
in_f <- "s12864-015-1767-y-s1.xlsx"
out_f <- "hoge3.txt"
param_skip <- 2
library(openxlsx)
data <- read.xlsx(in_f, startRow=param_skip, colNames=T, rowNames=T)
head(data)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
イントロ | 一般 | ExpressionSet | 1から作成 | Biobase
Biobaseパッケージ中のExpressionSet関数を用いて、
読み込んだ発現行列データをExpressionSet形式にするやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題6実行結果ファイルと同じものです。
3 female samples (HSF1-3) vs. 3 male samples (HSM1-3)の2群間比較用データです。
in_f <- "srp001558_count_hoge6.txt"
library(Biobase)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
eset <- ExpressionSet(assayData=as.matrix(data))
eset
例題1と基本的に同じですが、phenoData情報のところを追加しています。
in_f <- "srp001558_count_hoge6.txt"
param_G1 <- 3
param_G2 <- 3
library(Biobase)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
labelinfo <- data.frame(gender=as.factor(data.cl))
rownames(labelinfo) <- colnames(data)
metadata <- data.frame(labelDescription="nandemoiiyo",
row.names="gender")
phenoData <- new("AnnotatedDataFrame",
data=labelinfo,
varMetadata=metadata)
phenoData
pData(phenoData)
eset <- ExpressionSet(assayData=as.matrix(data),
phenoData=phenoData)
eset
イントロ | 一般 | ExpressionSet | 1から作成 | NOISeq(Tarazona_2015)
NOISeqパッケージ中のreadData関数を用いて、
読み込んだ発現行列データをExpressionSet形式にするやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題6実行結果ファイルと同じものです。
3 female samples (HSF1-3) vs. 3 male samples (HSM1-3)の2群間比較用データです。
「イントロ | 一般 | ExpressionSet | 1から作成 | Biobase」の例題2と同様の結果が得られていることが分かります。
in_f <- "srp001558_count_hoge6.txt"
param_G1 <- 3
param_G2 <- 3
library(NOISeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
myfactors <- data.frame(comp=data.cl)
eset <- readData(data=data, factors=myfactors)
eset
NGS機器(プラットフォーム)もいくつかあります:
イントロ | NGS | qPCRやmicroarrayなどとの比較
一言でいえば、3者の再現性は高いようですが、全般的にはRNA-seqのほうがダイナミックレンジが広く特に低発現領域で優れているようです。
NGS(RNA-seq) vs. RT-PCR:
- 't Hoen et al., Nucleic Acids Res., 2008:Figure 4
- Mane et al., BMC Genomics, 2009:Figure 3b, MAQCサンプルの論文
- Asmann et al., BMC Genomics, 2009:Figure 4
- Griffith et al., Nat. Methods, 2010:Figure 2, NACの論文;
- Lee et al., Nucleic Acids Res., 2010:Figure2と3, FVKMの論文
- Patro et al., Nat Biotechnol., 2014:Figure2a, Sailfishの論文
NGS(RNA-seq) vs. マイクロアレイ:
- Mortazavi et al., Nat Methods, 2008:Figure 3, RPKMの論文
- Sultan et al., Science, 2008:Figure 2, virtual lengthの論文
- Marioni et al., Genome Res., 2008:Figure 3と4
- 't Hoen et al., Nucleic Acids Res., 2008:Figure 3
- Mane et al., BMC Genomics, 2009:Supplementary Figureの下半分, MAQCサンプルの論文
- Richard et al., Nucleic Acids Res., 2010:Supplementary Figure S8, Solasの論文
- Griffith et al., Nat. Methods, 2010:Supplementary Figure 9aとb, NACの論文
- Nookaew et al., Nucleic Acids Res., 2012:Figure 2
- Kogenaru et al., BMC Genomics, 2012:Figure 1
- Sîrbu et al., PLoS One, 2012:Figure 2
- Xu et al., BMC Bioinformatics, 2013:Figure 2
- Guo et al., PLoS One, 2013:Figure 4
- Zhao et al., PLoS One, 2014:Figure 2
- Peng et al., PLoS One, 2014:Figure 1
- Wang et al., Nat Biotechnol., 2014:Figure 4
イントロ | NGS | 可視化(ゲノムブラウザやViewer)
可視化ツールも結構あります。
R用:
- TileQC:Dolan and Denver, BMC Bioinformatics, 2008
- GenomeGraphs:Durinck et al., BMC Bioinformatics, 2009
- HilbertVis:Anders S., Bioinformatics, 2009
- rtracklayer:Lawrence et al., Bioinformatics, 2009
- genoPlotR:Guy et al., Bioinformatics, 2010
- ggbio:Yin et al., Genome Biol., 2012
- rbamtools:Kaisers et al., Bioinformatics, 2015
- GenVisR:Skidmore et al., Bioinformatics, 2016
R以外(ウェブベースのゲノムブラウザ; server-side):
Review(Wang et al., Brief Bioinform., 2013)によるserver-sideと
client-sideの分類分けのうち、server-sideに相当するものたちだと思われます。
- VISTA:Frazer et al., Nucleic Acids Res., 2004
- Genome Projector(バクテリア系):Arakawa et al., BMC Bioinformatics, 2009
- Gramene(植物系):Jaiswal P, Methods Mol Biol., 2011
- Phytozome(植物系):Goodstein et al., Nucleic Acids Res., 2012
- The UCSC Genome Browser:Kuhn et al., Brief. Bioinform., 2013
- Ensembl:Zerbino et al., Nucleic Acids Res., 2018
- ChromoZoom:Pak et al., Bioinformatics, 2013
- Genome Maps:Medina et al., Nucleic Acids Res., 2013
- CCDS:Farrell et al., Nucleic Acids Res., 2014
R以外(stand-alone系):
- EagleView:Huang and Marth, Genome Res., 2008 (Linux, Windows, and Mac)
- MagicViewer:Hou et al., Nucleic Acids Res., 2010 (Linux, Windows, and Mac)
- MapView:Bao et al., Bioinformatics, 2009 (Linux and Windows)
- NGSView:Arner et al., Bioinformatics, 2010 (Linux)
- ZOOM Lite:Zhang et al., Nucleic Acids Res., 2010 (Linux and Windows)
- Tablet:Milne et al., Brief Bioinform., 2013 (Linux, Windows, and Mac)
- IGV:Thorvaldsdóttir et al., Brief. Bioinform., 2013 (Linux, Windows, and Mac)
- GenomeVISTA:Poliakov et al., Bioinformatics, 2014
- bio-samtools 2:Etherington et al., Bioinformatics, 2015
イントロ | NGS | 配列取得 | FASTQ or SRA | 公共DBから
次世代シーケンサ(NGS)から得られる塩基配列データを公共データベースから取得する際には以下を利用します。
マイクロアレイデータ取得のときと同様、NGSデータもArrayExpress経由でダウンロードするのがいいかもしれません。
メタデータの全貌を把握しやすいこと、生データ(raw data)だけでなく加工済みのデータ(processed data)がある場合にはその存在がすぐにわかることなど、
操作性の点で他を凌駕していると思います。上記でも触れているようにFASTQファイルのダウンロードからマッピングまでを行うのはエンドユーザレベルでは大変ですが、
submitterが提供してくれている場合は(まだまだ少ないようですが)リファレンス配列へのマップ後のデータ、つまりBAM形式ファイルの提供もすでに始まっているようです。
2014年6月26日に知りました(DDBJ児玉さんありがとうございますm(_ _)m)。
データの形式は基本的にSanger typeのFASTQ形式です。
FASTA形式はリードあたり二行(idの行と配列の行)で表現します。
FASTQ形式はリードあたり4行(@から始まるidの行と配列の行、および+から始まるidの行とbase callの際のqualityの行)で表現します。
FASTQ形式は、Sangerのものがデファクトスタンダード(業界標準)です。
かつてIlluminaのプラットフォームから得られるのはFASTQ-like formatという表現がなされていたようです(Cock et al., Nucleic Acids Res., 2010)。
しかし少なくとも2013年頃には、IlluminaデータもBaseSpaceやCASAVA1.8のconfigureBclToFastq.plなどを用いることで業界標準のFASTQ形式(つまりSanger typeのデータ)に切り替えられるようですし、
NCBI SRAなどの公共DBから取得するデータは全てはSanger typeのデータになっていたと思います(Kibukawa E., テクニカルサポートウェビナー, 2013)。
- DDBJ Sequence Read Archive (DRA):Kodama et al., Nucleic Acids Res., 2018
- EMBL-EBI European Nucleotide Archive (ENA):Toribio et al., Nucleic Acids Res., 2017
- NCBI Sequence Read Archive (SRA):Sayers et al., Nucleic Acids Res., 2019
- ArrayExpress:Kolesnikov et al., Nucleic Acids Res., 2015
- GEO:Clough and Barrett, Methods Mol Biol., 2016
- DBCLS SRA:Nakazato et al., PLoS One, 2013
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)
SRAdbパッケージを用いてRNA-seq配列を取得するやり方を示します。
SRA形式ファイルの場合はNCBIからダウンロードしているようですが、FASTQ形式ファイルの場合はEBIからダウンロードしているようです(2014年6月23日、孫建強氏提供情報)。
ここではFASTQファイルをダウンロードするやり方を示します。2015年2月23日までオブジェクトkの中身の一部を取り出して表示させていましたが、
列名などが変わって取り出せなくなっていたのでこの際削除しました。またSRAmetadb.sqliteファイルは現在10GB超になっているようです。(2015年2月24日)。
getFASTQinfoとgetFASTQfile関数の部分にsra_con=sra_conを追加してエラーが出ないようにしました(2019年2月01日)。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
論文中の記述から腎臓(kidney: SRS000561)と肝臓(liver: SRS000562)サンプルの二群間比較であることがわかったうえで下記情報を眺めるとより理解が深まります。
この2のサンプルはさらに濃度別に2つに分類されています:腎臓3.0pM (SRX000605), 肝臓3.0pM (SRX000571), 腎臓1.5pM (SRX000606), 肝臓1.5pM (SRX000604)。
param <- "SRA000299"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run, sra_con=sra_con)
計6ファイル、合計7.3Gb程度の容量のファイルがダウンロードされます。東大の有線LANで一時間弱程度かかります。
早く終わらせたい場合は、最後のgetFASTQfile関数のオプションを'ftp'から'fasp'に変更すると時間短縮可能です。
param <- "SRA000299"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run, sra_con=sra_con)
#getSRAfile(hoge$run, srcType='ftp', fileType='fastq')
getFASTQfile(hoge$run, sra_con=sra_con, srcType='ftp')
論文中の記述からGSE42213を頼りに、
RNA-seqデータがGSE42212として収められていることを見出し、
その情報からSRP017142にたどり着いています。
計6ファイル、合計6Gb程度の容量のファイルがダウンロードされます。東大の有線LANで一時間弱程度かかります。
早く終わらせたい場合は、最後のgetFASTQfile関数のオプションを'ftp'から'fasp'に変更すると時間短縮可能です。
param <- "SRP017142"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run, sra_con=sra_con)
getFASTQfile(hoge$run, sra_con=sra_con, srcType='ftp')
4. シロイヌナズナのRNA-seqデータ("SRP011435":Huang et al., Development, 2012)のgzip圧縮済みのFASTQファイルをダウンロードする場合:
論文中の記述からGSE36469を頼りに、
RNA-seqデータがGSE36469として収められていることを見出し、
その情報からSRP011435にたどり着いています。
計8ファイル、合計10Gb程度の容量のファイルがダウンロードされます。東大の有線LANで2時間程度かかります。
param <- "SRP011435"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run, sra_con=sra_con)
getFASTQfile(hoge$run, sra_con=sra_con, srcType='ftp')
5. ニワトリのRNA-seqデータ("SRP038897":Sharon et al., PLoS One, 2014)のgzip圧縮済みのFASTQファイルをダウンロードする場合:
PacBioのデータです。計xファイル、合計x Gb程度の容量のファイルがダウンロードされます。東大の有線LANでx時間程度かかります。
param <- "SRP038897"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run, sra_con=sra_con)
getFASTQfile(hoge$run, sra_con=sra_con, srcType='ftp')
Illumina HiSeq 2000のpaired-endデータです。計6ファイル、合計8 Gb程度の容量のファイルがダウンロードされます。東大の有線LANで2時間程度かかります。
param <- "SRP017580"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run, sra_con=sra_con)
getFASTQfile(hoge$run, sra_con=sra_con, srcType='ftp')
7. カイコのsmall RNA-seqデータ("SRP016842":Nie et al., BMC Genomics, 2013)のgzip圧縮済みのFASTQファイルをダウンロードする場合:
論文中の記述からGSE41841を頼りに、
SRP016842にたどり着いています。
したがって、ここで指定するのは"SRP016842"となります。
以下を実行して得られるsmall RNA-seqファイルは1つ(SRR609266.fastq.gz)で、ファイルサイズは400Mb弱、11928428リードであることがわかります。
param <- "SRP016842"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run, sra_con=sra_con)
getFASTQfile(hoge$run, sra_con=sra_con, srcType='ftp')
イントロ | NGS | 配列取得 | シミュレーションデータ | について
NGSのシミュレーションリードデータを作成するプログラムもいくつかあるようです。ここで生成するのは塩基配列データです。
Shcherbina A., BMC Res Notes, 2014のTable 1にin silico read simulatorのまとめがあります。
R以外:
- MAQ(Illumina Solexa用; SNP系):Li et al., Genome Res., 2008
- MetaSim(454, Illumina, and Sanger用; メタゲノム系):Richter et al., PLoS One, 2008
- FlowSim(Roche 454用):Balzer et al., Bioinformatics, 2010
- DWGSIM(Illumina and IonTorrent用):原著論文はまだ
- ART(Roche 454 and Illumina Solexa用):Huang et al., Bioinformatics, 2012
- GemSIM(454 and Illumina用):McElroy et al., BMC Genomics, 2012
- Grinder(Platform-independent):Angly et al., Nucleic Acids Res., 2012
- pIRS(Illumina paired-end系):Hu et al., Bioinformatics, 2012
- flux simulator:Griebel et al., Nucleic Acids Res., 2012
- PBSIM(PacBio系):Ono et al., Bioinformatics, 2013
- NeSSM(メタゲノム系):Jia et al., PLoS One, 2013
- SInC(Snp, Indel and Cnv系):Pattnaik et al., BMC Bioinformatics, 2014
- FASTQSim:Shcherbina A., BMC Res Notes, 2014
イントロ | NGS | 配列取得 | シミュレーションデータ | ランダムな塩基配列の生成から
イントロ | 一般 | ランダムな塩基配列を生成と同じ手順でリファレンスとなる塩基配列を生成してから、
指定した配列長からなる部分配列を指定したリード数だけランダム抽出してシミュレーションNGSデータを生成するやり方を示します。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 50塩基の長さのリファレンス配列を生成したのち、20塩基長の部分配列を10リード分だけランダム抽出してFASTA形式で保存したい場合:
塩基の存在比はAが22%, Cが28%, Gが28%, Tが22%にしています。
out_f <- "hoge1.fasta"
param_len_ref <- 50
narabi <- c("A","C","G","T")
param_composition <- c(22, 28, 28, 22)
param_len_ngs <- 20
param_num_ngs <- 10
param_desc <- "kkk"
library(Biostrings)
set.seed(1010)
ACGTset <- rep(narabi, param_composition)
reference <- paste(sample(ACGTset, param_len_ref, replace=T), collapse="")
reference <- DNAStringSet(reference)
names(reference) <- param_desc
reference
s_posi <- sample(1:(param_len_ref-param_len_ngs), param_num_ngs, replace=T)
s_posi
hoge <- NULL
for(i in 1:length(s_posi)){
hoge <- append(hoge, subseq(reference, start=s_posi[i], width=param_len_ngs))
}
fasta <- hoge
description <- paste(param_desc, s_posi, (s_posi+param_len_ngs), sep="_")
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | NGS | アノテーション情報取得 | について
アノテーション情報の取り扱いやGFF形式のアノテーションファイルの読み込みなどは下記パッケージで利用可能です。例えば、
genomeIntervals中のreadGff3関数や、
GenomicFeatures中のmakeTxDbFromGFF関数などです。
実用上は、例えばbiomaRtはEnsembl gene IDのgene symbolを取得できるので、
Ensembl gene IDsをgene symbolsに変換するための基礎情報取得目的でも使えます。
- genomeIntervals
- rtracklayer(importやimport.gff関数):Lawrence et al., Bioinformatics, 2009
- biomaRt:Durinck et al., Nat Protoc., 2009
- easyRNASeq:Delhomme et al., Bioinformatics, 2012
- GenomicFeatures:Lawrence et al., PLoS Comput. Biol., 2013
- biomartr:Drost and Paszkowski, Bioinformatics, 2017
- ensembldb:Rainer et al., Bioinformatics, 2019
イントロ | NGS | アノテーション情報取得 | refFlat形式ファイル
数多くの生物種についてはUCSCのSequence and Annotation Downloadsから辿っていけばrefFlat形式の遺伝子アノテーションファイルを得ることができます。
このrefFlat形式のアノテーションファイルは、どの領域にどの遺伝子があるのかという座標(Coordinates)情報を含みます。
マイクロアレイ解析に慣れた人への説明としては、NGS(RNA-seqリード)のゲノムマッピング結果から遺伝子発現行列をお手軽に得るために必要なおまじないのファイル、のようなものだと解釈していただければいいと思います。
例として、ヒトとラットのrefFlatファイルを取得する手順を示します。
イントロ | NGS | アノテーション情報取得 | biomaRt(Durinck_2009)
biomaRtというRパッケージ(Durinck et al., Nat Protoc., 2009)を用いて、
様々な生物種のアノテーション情報を取得するやり方を示します。一般的にはBioMart (Kasprzyk A., Database, 2011)本体のウェブページ上でやるのでしょうが、私はGUIの変遷についていけませんorz...
ここでは、Ensembl Geneにデータセットを固定した状態で様々な生物種のアノテーション情報を取得するやり方を示します。
尚、Ensembl (Flicek et al., 2013)の遺伝子モデル(gene model)はリファレンスゲノムから直接アノテーションされたものであるのに対して、RefSeqはmRNA配列からアノテーションされたものです。
当然、リファレンスゲノムと個々のmRNA配列は異なりますので、RefSeq mRNA配列の一部はリファレンスゲノム配列にマップされません。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. ラット(Rattus norvegicus; rnorvegicus_gene_ensembl)の場合:
取得する情報を4つ指定しています: (1) "ensembl_gene_id", (2) "ensembl_transcript_id",
(3) "affy_rat230_2" (Affymetrix Rat Genome 230 2.0 Array; GPL1355のプローブセットID),
(4) "go_id"。
out_f <- "hoge1.txt"
param_dataset <- "rnorvegicus_gene_ensembl"
param_attribute <- c(
"ensembl_gene_id",
"ensembl_transcript_id",
"affy_rat230_2",
"go_id"
)
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
listAttributes(mart)
out <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(out)
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
2. ヒト(Homo Sapiens; hsapiens_gene_ensembl)の場合:
取得する情報を3つ指定しています: (1) "ensembl_transcript_id",
(2) "affy_hg_u133_plus_2" (Affymetrix Human Genome U133 Plus 2.0 Array; GPL570のプローブセットID),
(3) "refseq_mrna"。
27万行程度のファイルが得られます。
out_f <- "hoge2.txt"
param_dataset <- "hsapiens_gene_ensembl"
param_attribute <- c(
"ensembl_transcript_id",
"affy_hg_u133_plus_2",
"refseq_mrna"
)
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
listAttributes(mart)
out <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(out)
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
3. ヒト(Homo Sapiens; hsapiens_gene_ensembl)の場合:
取得する情報を3つ指定しています: (1) "refseq_mrna", (2) "hgnc_symbol",
(3) "go_id"。
56万行程度のファイルが得られます。
out_f <- "hoge3.txt"
param_dataset <- "hsapiens_gene_ensembl"
param_attribute <- c(
"refseq_mrna",
"hgnc_symbol",
"go_id"
)
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
listAttributes(mart)
out <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(out)
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
4. ヒト(Homo Sapiens; hsapiens_gene_ensembl)の場合:
取得する情報を3つ指定しています: (1) "refseq_mrna", (2) "hgnc_symbol",
(3) "go_id"。
3.と基本的には同じですが、refseq_mrna IDが存在するもののみフィルタリングした結果を出力しています。
42.6万行程度のファイルが得られます。以下では"with_ox_refseq_mrna"をオプションとして与えていますが、
なぜかlistFilters(mart)でリストアップされているので指定可能なはずの"refseq_mrna"でやるとエラーが出てしまいます。。。
out_f <- "hoge4.txt"
param_dataset <- "hsapiens_gene_ensembl"
param_attribute <- c(
"refseq_mrna",
"hgnc_symbol",
"go_id"
)
param_filter <- "with_ox_refseq_mrna"
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
listAttributes(mart)
out <- getBM(attributes=param_attribute, filters=param_filter, values=list(TRUE), mart=mart)
head(out)
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
5. ヒト(Homo Sapiens; hsapiens_gene_ensembl)の場合:
取得する情報を3つ指定しています: (1) "refseq_mrna", (2) "hgnc_symbol",
(3) "go_id"。
4.と同じ結果を得るべく、「refseq_mrna IDが存在するもののみフィルタリング」のところをgetBM関数のfiltersオプション使用以外の手段で行っています。
out_f <- "hoge5.txt"
param_dataset <- "hsapiens_gene_ensembl"
param_attribute <- c(
"refseq_mrna",
"hgnc_symbol",
"go_id"
)
library(biomaRt)
mart <- useMart("ensembl",dataset=param_dataset)
listAttributes(mart)
out <- getBM(attributes=param_attribute, filters="", values="", mart=mart)
head(out)
dim(out)
obj <- out[,1] != ""
out <- out[obj,]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
イントロ | NGS | アノテーション情報取得 | TxDb | について
アノテーション情報を取り扱うためにRでよく利用されるオブジェクトは、TxDb以外に、RangedData、GRangesListなどなどが挙げられますが、
このウェブページでは、TxDbというオブジェクトをアノテーション情報の基本オブジェクトとし、txdbというオブジェクト名で統一して取り扱っています。
このtxdbに対して、GenomicFeaturesで利用可能な
transcripts, exons, cds, genes, promoters, disjointExons, microRNAs, tRNAsなどの関数を適用して、GRanges形式のオブジェクトを得ることができます。
出力情報を制限したい場合には、 transcriptsByOverlaps, exonsByOverlaps, cdsByOverlapsなどの関数が利用可能です。
GRanges形式やGRangesList形式オブジェクトの取り扱いはGenomicRangesに記載されています。
イントロ | NGS | アノテーション情報取得 | TxDb | TxDb.*から
QuasRパッケージを用いてゲノムへのマッピング結果からカウント情報を得たいときに、
"TxDb"という形式のオブジェクトを利用する必要があります。TxDbオブジェクトは、makeTxDbFromGFF関数を用いてGTF形式ファイルを入力として作成することも可能ですが、
最も手っ取り早いやり方はTxDbオブジェクト形式で格納されている"TxDb.*"という名前のパッケージを利用することです。
利用可能なTxDb.*パッケージはここにリストアップされているものたちです。
(1)「全パッケージリスト(All Packages)」中の、
(2)「AnnotationDataの左側のさんかく」、
(3)「PackageTypeの左側のさんかく」、
(4)「TxDb」からも辿れます。
ここでは、いくつかのパッケージの読み込みまでを示します。
1. TxDb.Hsapiens.UCSC.hg19.knownGene(ヒト)の場合:
hg19 (Genome Reference Consortium GRCh37のことらしい)です。
param <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
txdb <- eval(parse(text=tmp))
txdb
2. TxDb.Rnorvegicus.UCSC.rn5.refGene(ラット)の場合:
2015年2月現在、Rで取得可能なラットの最新版です。
param <- "TxDb.Rnorvegicus.UCSC.rn5.refGene"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
txdb <- eval(parse(text=tmp))
txdb
3. TxDb.Hsapiens.UCSC.hg38.knownGene(ヒト)の場合:
hg38 (Genome Reference Consortium GRCh38のことらしい)です。
param <- "TxDb.Hsapiens.UCSC.hg38.knownGene"
library(param, character.only=T)
#tmp <- unlist(strsplit(param, ".", fixed=TRUE))[2]
tmp <- ls(paste("package", param, sep=":"))
txdb <- eval(parse(text=tmp))
txdb
イントロ | NGS | アノテーション情報取得 | TxDb | GenomicFeatures(Lawrence_2013)
QuasRパッケージを用いてゲノムへのマッピング結果からカウント情報を得たいときに、
"TxDb"という形式のオブジェクトを利用する必要があります。ここでは、GenomicFeatures
パッケージを用いてTxDbオブジェクトを得るやり方を示します。
得られるTxDbオブジェクトの元データのバージョンは、当然この項目で作成されたもののほうが新しいです。
したがって、慣れてきたら、予め作成された"TxDb.*"パッケージのものを使うよりもここで直接作成してしまうほうがいいでしょう。
しかし、逆に言えば作成するたびに転写物数("transcript_nrow"のところの数値など)が異なってきますので、"RSQLite version at creation time"のところの情報は気にしたほうがいいでしょう。
どういうものを取得可能かリストアップしているだけです。
library(GenomicFeatures)
library(rtracklayer)
ucscGenomes()
supportedUCSCtables()
ヒトゲノム ver. 19 ("hg19"; Genome Reference Consortium GRCh37のことらしい)のUCSC Genes ("knownGene")のTxDbオブジェクトを取得するやり方です。
実質的に、"TxDb.Hsapiens.UCSC.hg19.knownGene"パッケージから取得可能なものと同じ(はず)です。
param1 <- "hg19"
param2 <- "knownGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
ヒトゲノム ver. 19 ("hg19")のEnsembl Genes ("ensGene")のTxDbオブジェクトを取得するやり方です。
IDはEntrez Gene IDのままで、配列長情報などがEnsembl Genesのものになるだけのようです。
param1 <- "hg19"
param2 <- "ensGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
transcripts(txdb)
4. Ensemblから遺伝子アノテーション情報を取得する場合:
ヒト("hsapiens_gene_ensembl")のTxDbオブジェクトを取得するやり方です。
param <- "hsapiens_gene_ensembl"
library(GenomicFeatures)
txdb <- makeTxDbFromBiomart(dataset=param)
txdb
transcripts(txdb)
マウスゲノム ("mm10"; Genome Reference Consortium GRCm38)のEnsembl Genes ("ensGene")のTxDbオブジェクトを取得するやり方です。
param1 <- "mm10"
param2 <- "ensGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
transcripts(txdb)
ラットゲノム ("rn5"; RGSC Rnor_5.0)のEnsembl Genes ("ensGene")のTxDbオブジェクトを取得するやり方です。
param1 <- "rn5"
param2 <- "ensGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
transcripts(txdb)
ヒツジゲノム ("oviAri3"; ISGC Oar_v3.1)のEnsembl Genes ("ensGene")のTxDbオブジェクトを取得するやり方です。
param1 <- "oviAri3"
param2 <- "ensGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
transcripts(txdb)
ニワトリゲノム ("galGal4"; ICGC Gallus-gallus-4.0)のEnsembl Genes ("ensGene")のTxDbオブジェクトを取得するやり方です。
param1 <- "galGal4"
param2 <- "ensGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
transcripts(txdb)
ヒトゲノム("hg38"; Genome Reference Consortium GRCh38のことらしい)のUCSC Genes ("knownGene")のTxDbオブジェクトを取得するやり方です。
param1 <- "hg38"
param2 <- "knownGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
ヒトゲノム("hg38"; Genome Reference Consortium GRCh38のことらしい)のEnsembl Genes ("ensGene")のTxDbオブジェクトを取得するやり方です。
エラーが出ます。
param1 <- "hg38"
param2 <- "ensGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
ヒトゲノム("hg38"; Genome Reference Consortium GRCh38のことらしい)のRefSeq Genes ("refGene")のTxDbオブジェクトを取得するやり方です。
param1 <- "hg38"
param2 <- "refGene"
library(GenomicFeatures)
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
txdb
イントロ | NGS | アノテーション情報取得 | TxDb | GFF/GTF形式ファイルから
QuasRパッケージを用いてゲノムへのマッピング結果からカウント情報を得たいときに、
"TxDb"という形式のオブジェクトを利用する必要があります。ここでは、GenomicFeatures
パッケージを用いて手元にあるGFF/GTF形式ファイルを入力としてTxDbオブジェクトを得るやり方を示します。
基本的にはGenomicFeaturesパッケージ中のmakeTxDbFromGFF関数を用いてGFF/GTF形式ファイルを読み込むことで
TxDbオブジェクトをエラーなく読み込むこと自体は簡単にできます。しかし、得られたTxDbオブジェクトとゲノムマッピング結果ファイルを用いてカウント情報を得る場合に、
ゲノム配列提供元とアノテーション情報提供元が異なっているとエラーとなります。具体的には、GFF/GTFファイル中にゲノム配列中にない染色体名があるとエラーが出る場合があります。
基本形です。エラーは出ませんが、2015年3月4日現在、ChrCが環状ではないと認識されてしまっています。
in_f <- "TAIR10_GFF3_genes.gff"
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f)
txdb
基本形です。2019年4月19日に動作確認した結果、エラーが出て読み込めなくなっているようです。
in_f <- "human_annotation_sub3.gtf"
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f, format="gtf")
txdb
うまくいく例です。
in_f1 <- "human_annotation_sub3.gtf"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
library(GenomicFeatures)
param <- in_f2
library(param, character.only=T)
tmp <- ls(paste("package", param, sep=":"))
hoge <- eval(parse(text=tmp))
chrominfo <- data.frame(chrom=as.character(seqnames(hoge)),
length=as.vector(seqlengths(hoge)),
is_circular=hoge@seqinfo@is_circular)
txdb <- makeTxDbFromGFF(file=in_f1, format="gtf",
chrominfo=chrominfo,
species=organism(hoge))
txdb
うまくいかない例です。
in_f1 <- "human_annotation_sub2.gtf"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
library(GenomicFeatures)
param <- in_f2
library(param, character.only=T)
tmp <- ls(paste("package", param, sep=":"))
hoge <- eval(parse(text=tmp))
chrominfo <- data.frame(chrom=as.character(seqnames(hoge)),
length=as.vector(seqlengths(hoge)),
is_circular=hoge@seqinfo@is_circular)
txdb <- makeTxDbFromGFF(file=in_f1, format="gtf",
chrominfo=chrominfo,
species=organism(hoge))
txdb
うまくいかない例です。
in_f1 <- "human_annotation_sub1.gtf"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
library(GenomicFeatures)
param <- in_f2
library(param, character.only=T)
tmp <- ls(paste("package", param, sep=":"))
hoge <- eval(parse(text=tmp))
chrominfo <- data.frame(chrom=as.character(seqnames(hoge)),
length=as.vector(seqlengths(hoge)),
is_circular=hoge@seqinfo@is_circular)
txdb <- makeTxDbFromGFF(file=in_f1, format="gtf",
exonRankAttributeName="exon_number",
gffGeneIdAttributeName="gene_name",
chrominfo=chrominfo,
species=organism(hoge))
txdb
TAIR(Lamesch et al., 2012)
から提供されているものです。
1.と基本的に同じですが、どの生物種に対してもデフォルトではChrMのみ環状ゲノムであるとしてしまうようですので、
circ_seqsオプションでChrCが環状であると認識させるやり方です(高橋 広夫 氏提供情報)。
in_f <- "TAIR10_GFF3_genes.gff"
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f, format="gff3",
circ_seqs=c("ChrC","ChrM"))
txdb
seqinfo(txdb)
- GenomicFeatures:Lawrence et al., PLoS Comput. Biol., 2013
- QuasR:Gaidatzis et al., Bioinformatics, 2015
- Ensembl:Zerbino et al., Nucleic Acids Res., 2018
- TAIR:Reiser et al., Curr Protoc Bioinformatics., 2017
- Lactobacillus hokkaidonensis JCM 18461:Tanizawa et al., BMC Genomics, 2015
イントロ | NGS | 読み込み | BSgenome | 基本情報を取得
BSgenomeパッケージを読み込んで、
Total lengthやaverage lengthなどの各種情報取得を行うためのやり方を示します。
パッケージがインストールされていない場合は、インストール | Rパッケージ | 個別
を参考にしてインストールしておく必要があります。
マウスやヒトゲノム解析の場合に「整数オーバーフロー」問題が生じていましたが、Total_lenのところで「sum(width(fasta))」を「sum(as.numeric(width(fasta)))」に、
そしてsortedのところで「rev(sort(width(fasta)))」を「rev(sort(as.numeric(width(fasta))))」などと書き換えることで回避可能であるという情報をいただきましたので、
2015年5月27日にそのように変更しました(野間口達洋 氏提供情報)。
「ファイル」−「ディレクトリの変更」で出力結果ファイルを保存したいディレクトリに移動し以下をコピペ。
GC含量は約41%となります。
これは、GとCが各20.5%を占め、残りのAとTが各29.5%を占めることを意味します。
out_f <- "hoge1.txt"
param_bsgenome <- "BSgenome.Hsapiens.NCBI.GRCh38"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ゲノムサイズが約120MB、GC含量が約36%であることがわかります。
これは、GとCが各18%を占め、残りのAとTが各32%を占めることを意味します。
out_f <- "hoge2.txt"
param_bsgenome <- "BSgenome.Athaliana.TAIR.TAIR9"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ゲノムサイズが約100MB、GC含量が約35.4%であることがわかります。
これは、GとCが各17.7%を占め、残りのAとTが各32.3%を占めることを意味します。
out_f <- "hoge3.txt"
param_bsgenome <- "BSgenome.Celegans.UCSC.ce6"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ゲノムサイズが約1.357GB、GC含量が約36.6%であることがわかります。
これは、GとCが各18.3%を占め、残りのAとTが各31.7%を占めることを意味します。
out_f <- "hoge4.txt"
param_bsgenome <- "BSgenome.Drerio.UCSC.danRer7"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
GC含量は約41.667%という値が得られています。
これは、GとCが各20.8335%を占め、残りのAとTが各29.1665%を占めることを意味します。
out_f <- "hoge5.txt"
param_bsgenome <- "BSgenome.Mmusculus.UCSC.mm10"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
GC含量は約50.631%という値が得られています。
これは、GとCが各25.316%を占め、残りのAとTが各24.684%を占めることを意味します。
out_f <- "hoge6.txt"
param_bsgenome <- "BSgenome.Ecoli.NCBI.20080805"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
GC含量は約41.057%という値が得られています。
これは、GとCが各20.528%を占め、残りのAとTが各29.472%を占めることを意味します。
out_f <- "hoge7.txt"
param_bsgenome <- "BSgenome.Cfamiliaris.UCSC.canFam3"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
GC含量は約42.21%という値が得られています。
これは、GとCが各21.11%を占め、残りのAとTが各28.89%を占めることを意味します。
out_f <- "hoge8.txt"
param_bsgenome <- "BSgenome.Dmelanogaster.UCSC.dm2"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
GC含量は約43.57%という値が得られています。
これは、GとCが各21.78%を占め、残りのAとTが各28.22%を占めることを意味します。
out_f <- "hoge9.txt"
param_bsgenome <- "BSgenome.Osativa.MSU.MSU7"
library(Biostrings)
library(param_bsgenome, character.only=T)
tmp <- ls(paste("package", param_bsgenome, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
イントロ | NGS | 読み込み | FASTA形式 | 基本情報を取得
multi-FASTAファイルを読み込んで、Total lengthやaverage lengthなどの各種情報取得を行うためのやり方を示します。
例題6以降は、ヒトやマウスレベルの巨大ファイルを取り扱うためのコードです。具体的には、
塩基数を整数(integer)ではなく実数(real number)として取り扱うためのas.numeric関数を追加しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "hoge4.fa"
out_f <- "hoge1.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
Total_len <- sum(width(fasta))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(width(fasta)))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(CG)/sum(ACGT)
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. 130MB程度のRefSeqのhuman mRNAのmulti-FASTAファイル(h_rna.fasta)の場合:
in_f <- "h_rna.fasta"
out_f <- "hoge2.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
Total_len <- sum(width(fasta))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(width(fasta)))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(CG)/sum(ACGT)
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
FASTA形式ファイルの読み込み部分で、Biostringsパッケージ中のreadDNAStringSet関数ではなく、seqinrパッケージ中のread.fasta関数を用いるやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge3.txt"
library(Biostrings)
library(seqinr)
hoge <- read.fasta(in_f, seqtype="DNA", as.string=TRUE)
fasta <- DNAStringSet(as.character(hoge))
names(fasta) <- names(hoge)
Total_len <- sum(width(fasta))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(width(fasta)))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(CG)/sum(ACGT)
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
4. 120MB程度のシロイヌナズナゲノムのmulti-FASTAファイル(TAIR10_chr_all.fas.gz)の場合:
in_f <- "TAIR10_chr_all.fas.gz"
out_f <- "hoge4.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
Total_len <- sum(width(fasta))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(width(fasta)))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(CG)/sum(ACGT)
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. Ensembl Genomesから取得した乳酸菌ゲノムのmulti-FASTAファイル(Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa)の場合:
Lactobacillus casei 12A (Taxonomy ID: 1051650):Broadbent et al., BMC Genomics, 2012のゲノム配列です。
Lactobacillus casei 12A - Download DNA sequence
- Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa.gzをダウンロード後に解凍すれば、同じファイル名のものが得られます。
in_f <- "Lactobacillus_casei_12a.GCA_000309565.1.22.dna.toplevel.fa"
out_f <- "hoge5.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
Total_len <- sum(width(fasta))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(width(fasta)))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(CG)/sum(ACGT)
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
6. ヒトやマウスレベルのmulti-FASTAファイルの場合:
例題5までのコードは、マウスやヒトゲノム解析の場合に「整数オーバーフロー」問題が生じてうまくいきませんが、それを回避するやり方です。
具体的には、Total_lenのところで「sum(width(fasta))」を「sum(as.numeric(width(fasta)))」に、
そしてsortedのところで「rev(sort(width(fasta)))」を「rev(sort(as.numeric(width(fasta))))」と書き換えています(野間口達洋 氏提供情報)。
in_f <- "hoge9.fa"
out_f <- "hoge6.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
Total_len <- sum(as.numeric(width(fasta)))
Number_of_contigs <- length(fasta)
Average_len <- mean(width(fasta))
Median_len <- median(width(fasta))
Max_len <- max(width(fasta))
Min_len <- min(width(fasta))
sorted <- rev(sort(as.numeric(width(fasta))))
obj <- (cumsum(sorted) >= Total_len*0.5)
N50 <- sorted[obj][1]
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- sum(as.numeric(CG))/sum(as.numeric(ACGT))
tmp <- NULL
tmp <- rbind(tmp, c("Total length (bp)", Total_len))
tmp <- rbind(tmp, c("Number of contigs", Number_of_contigs))
tmp <- rbind(tmp, c("Average length", Average_len))
tmp <- rbind(tmp, c("Median length", Median_len))
tmp <- rbind(tmp, c("Max length", Max_len))
tmp <- rbind(tmp, c("Min length", Min_len))
tmp <- rbind(tmp, c("N50", N50))
tmp <- rbind(tmp, c("GC content", GC_content))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
イントロ | NGS | 読み込み | FASTA形式 | description行の記述を整形
multi-FASTAファイルのdescription行の記述はものによってまちまちです。ここでは何種類かのmulti-FASTA形式ファイルに対して、
「目的の記述部分のみ抽出し、それを新たなdescription」とするやり方や、まるごと指定した文字列に置換するやり方などを示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. ラットの上流配列(1000bp)ファイル(rat_upstream_1000.fa)からRefSeq IDの部分のみを抽出したい場合:
抽出例:「NM_022953_up_1000_chr1_268445091_r chr1:268445091-268446090」--> 「NM_022953」
戦略:"_up_"を区切り文字として分割("NM_022953"と"1000_chr1_268445091_r chr1:268445091-268446090")し、分割後の1つ目の要素を抽出
例題ファイルのダウンロード時に、なぜか拡張子の.faが.txtに勝手に変更されていたりする場合がありますのでご注意ください。
in_f <- "rat_upstream_1000.fa"
out_f <- "hoge1.fasta"
param1 <- "_up_"
param2 <- 1
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- strsplit(names(fasta), param1, fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", param2))
names(fasta) <- hoge2
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. Trinity.fastaから最初のスペースで区切られる前の文字列のみにしたい場合:
抽出例:「comp59_c0_seq1 len=537 ~FPKM=305.1 path=[0:0-536]」--> 「comp59_c0_seq1」
戦略:" "を区切り文字として分割("comp59_c0_seq1", "len=537", "~FPKM=305.1", "path=[0:0-536]")し、分割後の1つ目の要素を抽出
in_f <- "Trinity.fasta"
out_f <- "hoge2.fasta"
param1 <- " "
param2 <- 1
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- strsplit(names(fasta), param1, fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", param2))
names(fasta) <- hoge2
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. DHFR.fastaからRefSeq ID部分のみ抽出したい場合:
抽出例:「gi|68303806|ref|NM_000791.3| Homo sapiens dihydrofolate reductase (DHFR), mRNA」--> 「NM_000791.3」
戦略:"|"を区切り文字として分割("gi", "68303806", "ref", "NM_000791.3", " Homo sapiens dihydrofolate reductase (DHFR), mRNA")し、分割後の4番目の要素を抽出
in_f <- "DHFR.fasta"
out_f <- "hoge3.fasta"
param1 <- "|"
param2 <- 4
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- strsplit(names(fasta), param1, fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", param2))
names(fasta) <- hoge2
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. DHFR.fastaからバージョン番号を除いたRefSeq ID部分のみ抽出したい場合:
抽出例:「gi|68303806|ref|NM_000791.3| Homo sapiens dihydrofolate reductase (DHFR), mRNA」--> 「NM_000791」
戦略:"|"を区切り文字として分割("gi", "68303806", "ref", "NM_000791.3", " Homo sapiens dihydrofolate reductase (DHFR), mRNA")し、分割後の4番目の要素を抽出。
次に、"."を区切り文字として分割("NM_000791", "3")し、分割後の1番目の要素を抽出。
in_f <- "DHFR.fasta"
out_f <- "hoge4.fasta"
param1 <- "|"
param2 <- 4
param3 <- "."
param4 <- 1
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- strsplit(names(fasta), param1, fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", param2))
hoge3 <- strsplit(hoge2, param3, fixed=TRUE)
hoge4 <- unlist(lapply(hoge3, "[[", param4))
names(fasta) <- hoge4
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
5. sample_100.fastaからバージョン番号を除いたRefSeq ID部分のみ抽出したい場合:
抽出例:「gi|239744030|ref|XR_017086.3| PREDICTED: Homo sapiens supervillin-like (LOC645954), miscRNA」--> 「XR_017086」
戦略:"|"を区切り文字として分割("gi", "239744030", "ref", "XR_017086.3", " PREDICTED: Homo sapiens supervillin-like (LOC645954), miscRNA")し、分割後の4番目の要素を抽出。
次に、"."を区切り文字として分割("XR_017086", "3")し、分割後の1番目の要素を抽出。
in_f <- "sample_100.fasta"
out_f <- "hoge5.fasta"
param1 <- "|"
param2 <- 4
param3 <- "."
param4 <- 1
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- strsplit(names(fasta), param1, fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", param2))
hoge3 <- strsplit(hoge2, param3, fixed=TRUE)
hoge4 <- unlist(lapply(hoge3, "[[", param4))
names(fasta) <- hoge4
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
染色体の並びがparamで指定したものと同じであることが既知という前提です。ゲノム配列ファイル自体は120MB程度あります。
in_f <- "TAIR10_chr_all.fas.gz"
out_f <- "hoge6.fasta"
param <- c("Chr1","Chr2","Chr3","Chr4","Chr5","ChrM","ChrC")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
names(fasta) <- param
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | NGS | 読み込み | FASTQ形式 | 基礎
Sanger FASTQ形式ファイルを読み込むやり方を示します。「基礎」では、FASTQファイルの中身を全て読み込む手順を示します。
入力、出力形式は、ともに非圧縮(.fastq)、gzip圧縮(.fastq.gz)ファイルが可能です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
quality情報を除く塩基配列情報のみ読み込むやり方です。配列長が同じ場合のみ読み込めます。
in_f <- "SRR037439.fastq"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
quality情報も読み込むやり方です。配列長が異なっていても読み込めます。
in_f <- "SRR037439.fastq"
library(ShortRead)
fastq <- readFastq(in_f)
fastq
showClass("ShortReadQ")
sread(fastq)
quality(fastq)
id(fastq)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
quality情報も読み込むやり方です。配列長が異なっていても読み込めます。
description行の" "以降の文字を削除して、またFASTQ形式で保存するやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge3.fastq"
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), " ", fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", 1))
fastq <- ShortReadQ(sread(fastq), quality(fastq), description)
id(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
quality情報も読み込むやり方です。配列長が異なっていても読み込めます。
description行の" "以降の文字を削除して、またFASTQ形式で保存するやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
in_f <- "SRR037439.fastq"
out_f <- "hoge4.fastq.gz"
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), " ", fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", 1))
fastq <- ShortReadQ(sread(fastq), quality(fastq), description)
id(fastq)
writeFastq(fastq, out_f, compress=T)
5. small RNA-seqのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られた
カイコsmall RNA-seqデータ(Nie et al., BMC Genomics, 2013)です。
入力ファイルサイズは400Mb弱、11,928,428リードです。
この中から100000リード分をランダムに非復元抽出した結果をgzip圧縮なしで出力しています。
出力ファイルはSRR609266_sub.fastqと同じもの(100000リード; 約16MB)になります。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge5.fastq"
param <- 100000
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
set.seed(1010)
obj <- sample(1:length(fastq), param, replace=F)
fastq <- fastq[sort(obj)]
id(fastq)
writeFastq(fastq, out_f, compress=F)
6. small RNA-seqのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られた
カイコsmall RNA-seqデータ(Nie et al., BMC Genomics, 2013)です。
入力ファイルサイズは400Mb弱、11,928,428リードです。この中から最初の100000リード分をgzip圧縮なしで出力しています。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge6.fastq"
param <- 100000
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
fastq <- fastq[1:param]
id(fastq)
writeFastq(fastq, out_f, compress=F)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約73MB)です。長さは全て107 bpです。
NAを含む場合への各種対応策を2015年6月24日に追加しました(茂木朋貴氏、野間口達洋氏、他多くの受講生提供情報)。
in_f <- "SRR616268sub_1.fastq.gz"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
hoge <- subseq(fasta, start=1, end=7)
hoge
head(table(hoge))
head(sort(table(hoge), decreasing=T))
table(hoge)["CGGGCCT"]
hoge <- subseq(fasta, start=3, width=7)
hoge
table(hoge)["CGGGCCT"]
hoge <- subseq(fasta, start=5, width=7)
table(hoge)["CGGGCCT"]
table(subseq(fasta, start=1, width=7))["CGGGCCT"]
table(subseq(fasta, start=2, width=7))["CGGGCCT"]
table(subseq(fasta, start=3, width=7))["CGGGCCT"]
table(subseq(fasta, start=4, width=7))["CGGGCCT"]
table(subseq(fasta, start=5, width=7))["CGGGCCT"]
table(subseq(fasta, start=6, width=7))["CGGGCCT"]
table(subseq(fasta, start=7, width=7))["CGGGCCT"]
table(subseq(fasta, start=8, width=7))["CGGGCCT"]
table(subseq(fasta, start=9, width=7))["CGGGCCT"]
table(subseq(fasta, start=10, width=7))["CGGGCCT"]
table(subseq(fasta, start=11, width=7))["CGGGCCT"]
table(subseq(fasta, start=12, width=7))["CGGGCCT"]
table(subseq(fasta, start=13, width=7))["CGGGCCT"]
table(subseq(fasta, start=14, width=7))["CGGGCCT"]
table(subseq(fasta, start=15, width=7))["CGGGCCT"]
table(subseq(fasta, start=16, width=7))["CGGGCCT"]
table(subseq(fasta, start=17, width=7))["CGGGCCT"]
table(subseq(fasta, start=18, width=7))["CGGGCCT"]
table(subseq(fasta, start=19, width=7))["CGGGCCT"]
table(subseq(fasta, start=20, width=7))["CGGGCCT"]
table(subseq(fasta, start=21, width=7))["CGGGCCT"]
table(subseq(fasta, start=22, width=7))["CGGGCCT"]
table(subseq(fasta, start=23, width=7))["CGGGCCT"]
table(subseq(fasta, start=24, width=7))["CGGGCCT"]
table(subseq(fasta, start=25, width=7))["CGGGCCT"]
table(subseq(fasta, start=26, width=7))["CGGGCCT"]
table(subseq(fasta, start=27, width=7))["CGGGCCT"]
table(subseq(fasta, start=28, width=7))["CGGGCCT"]
table(subseq(fasta, start=29, width=7))["CGGGCCT"]
table(subseq(fasta, start=30, width=7))["CGGGCCT"]
table(subseq(fasta, start=31, width=7))["CGGGCCT"]
table(subseq(fasta, start=32, width=7))["CGGGCCT"]
table(subseq(fasta, start=33, width=7))["CGGGCCT"]
table(subseq(fasta, start=34, width=7))["CGGGCCT"]
table(subseq(fasta, start=35, width=7))["CGGGCCT"]
table(subseq(fasta, start=36, width=7))["CGGGCCT"]
table(subseq(fasta, start=37, width=7))["CGGGCCT"]
table(subseq(fasta, start=38, width=7))["CGGGCCT"]
table(subseq(fasta, start=39, width=7))["CGGGCCT"]
table(subseq(fasta, start=40, width=7))["CGGGCCT"]
table(subseq(fasta, start=41, width=7))["CGGGCCT"]
table(subseq(fasta, start=42, width=7))["CGGGCCT"]
table(subseq(fasta, start=43, width=7))["CGGGCCT"]
table(subseq(fasta, start=44, width=7))["CGGGCCT"]
table(subseq(fasta, start=45, width=7))["CGGGCCT"]
table(subseq(fasta, start=46, width=7))["CGGGCCT"]
table(subseq(fasta, start=47, width=7))["CGGGCCT"]
table(subseq(fasta, start=48, width=7))["CGGGCCT"]
table(subseq(fasta, start=49, width=7))["CGGGCCT"]
table(subseq(fasta, start=50, width=7))["CGGGCCT"]
table(subseq(fasta, start=51, width=7))["CGGGCCT"]
table(subseq(fasta, start=52, width=7))["CGGGCCT"]
table(subseq(fasta, start=53, width=7))["CGGGCCT"]
table(subseq(fasta, start=54, width=7))["CGGGCCT"]
table(subseq(fasta, start=55, width=7))["CGGGCCT"]
table(subseq(fasta, start=56, width=7))["CGGGCCT"]
table(subseq(fasta, start=57, width=7))["CGGGCCT"]
table(subseq(fasta, start=58, width=7))["CGGGCCT"]
table(subseq(fasta, start=59, width=7))["CGGGCCT"]
table(subseq(fasta, start=60, width=7))["CGGGCCT"]
table(subseq(fasta, start=61, width=7))["CGGGCCT"]
table(subseq(fasta, start=62, width=7))["CGGGCCT"]
table(subseq(fasta, start=63, width=7))["CGGGCCT"]
table(subseq(fasta, start=64, width=7))["CGGGCCT"]
table(subseq(fasta, start=65, width=7))["CGGGCCT"]
table(subseq(fasta, start=66, width=7))["CGGGCCT"]
table(subseq(fasta, start=67, width=7))["CGGGCCT"]
table(subseq(fasta, start=68, width=7))["CGGGCCT"]
table(subseq(fasta, start=69, width=7))["CGGGCCT"]
table(subseq(fasta, start=70, width=7))["CGGGCCT"]
table(subseq(fasta, start=71, width=7))["CGGGCCT"]
table(subseq(fasta, start=72, width=7))["CGGGCCT"]
table(subseq(fasta, start=73, width=7))["CGGGCCT"]
table(subseq(fasta, start=74, width=7))["CGGGCCT"]
table(subseq(fasta, start=75, width=7))["CGGGCCT"]
table(subseq(fasta, start=76, width=7))["CGGGCCT"]
table(subseq(fasta, start=77, width=7))["CGGGCCT"]
table(subseq(fasta, start=78, width=7))["CGGGCCT"]
table(subseq(fasta, start=79, width=7))["CGGGCCT"]
table(subseq(fasta, start=80, width=7))["CGGGCCT"]
table(subseq(fasta, start=81, width=7))["CGGGCCT"]
table(subseq(fasta, start=82, width=7))["CGGGCCT"]
table(subseq(fasta, start=83, width=7))["CGGGCCT"]
table(subseq(fasta, start=84, width=7))["CGGGCCT"]
table(subseq(fasta, start=85, width=7))["CGGGCCT"]
table(subseq(fasta, start=86, width=7))["CGGGCCT"]
table(subseq(fasta, start=87, width=7))["CGGGCCT"]
table(subseq(fasta, start=88, width=7))["CGGGCCT"]
table(subseq(fasta, start=89, width=7))["CGGGCCT"]
table(subseq(fasta, start=90, width=7))["CGGGCCT"]
table(subseq(fasta, start=91, width=7))["CGGGCCT"]
table(subseq(fasta, start=92, width=7))["CGGGCCT"]
table(subseq(fasta, start=93, width=7))["CGGGCCT"]
table(subseq(fasta, start=94, width=7))["CGGGCCT"]
table(subseq(fasta, start=95, width=7))["CGGGCCT"]
table(subseq(fasta, start=96, width=7))["CGGGCCT"]
table(subseq(fasta, start=97, width=7))["CGGGCCT"]
table(subseq(fasta, start=98, width=7))["CGGGCCT"]
table(subseq(fasta, start=99, width=7))["CGGGCCT"]
table(subseq(fasta, start=100, width=7))["CGGGCCT"]
table(subseq(fasta, start=101, width=7))["CGGGCCT"]
param_kmer <- 7
param_obj <- "CGGGCCT"
Obs <- NULL
for(i in 1:101){
Obs <- c(Obs, table(subseq(fasta, start=i, width=param_kmer))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
param_obj <- "CGGGCCT"
Obs <- NULL
for(i in 1:101){
Obs <- c(Obs, table(subseq(fasta, start=i, width=nchar(param_obj)))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
param_len_ngs <- 107
param_obj <- "CGGGCCT"
Obs <- NULL
hoge <- param_len_ngs - nchar(param_obj) + 1
for(i in 1:hoge){
Obs <- c(Obs, table(subseq(fasta, start=i, width=nchar(param_obj)))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
param_obj <- "CGGGCCT"
Obs <- NULL
hoge <- width(fasta)[1] - nchar(param_obj) + 1
for(i in 1:hoge){
Obs <- c(Obs, table(subseq(fasta, start=i, width=nchar(param_obj)))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約73MB)です。長さは全て107 bpです。
NAを含む場合への各種対応策を2015年6月24日に追加しました(茂木朋貴氏、野間口達洋氏、他多くの受講生提供情報)。
8.と基本的に同じですが、入力ファイルの12%分(120,000リード)のみで解析するとFastQC実行結果と同じになることがわかったので、そのサブセットのみで再解析しています。/p>
in_f <- "SRR616268sub_1.fastq.gz"
param <- 120000
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
fasta <- fasta[1:param]
fasta
hoge <- subseq(fasta, start=1, end=7)
hoge
head(table(hoge))
head(sort(table(hoge), decreasing=T))
table(hoge)["CGGGCCT"]
hoge <- subseq(fasta, start=3, width=7)
hoge
table(hoge)["CGGGCCT"]
hoge <- subseq(fasta, start=5, width=7)
table(hoge)["CGGGCCT"]
table(subseq(fasta, start=1, width=7))["CGGGCCT"]
table(subseq(fasta, start=2, width=7))["CGGGCCT"]
table(subseq(fasta, start=3, width=7))["CGGGCCT"]
table(subseq(fasta, start=4, width=7))["CGGGCCT"]
table(subseq(fasta, start=5, width=7))["CGGGCCT"]
table(subseq(fasta, start=6, width=7))["CGGGCCT"]
table(subseq(fasta, start=7, width=7))["CGGGCCT"]
table(subseq(fasta, start=8, width=7))["CGGGCCT"]
table(subseq(fasta, start=9, width=7))["CGGGCCT"]
table(subseq(fasta, start=10, width=7))["CGGGCCT"]
table(subseq(fasta, start=11, width=7))["CGGGCCT"]
table(subseq(fasta, start=12, width=7))["CGGGCCT"]
table(subseq(fasta, start=13, width=7))["CGGGCCT"]
table(subseq(fasta, start=14, width=7))["CGGGCCT"]
table(subseq(fasta, start=15, width=7))["CGGGCCT"]
table(subseq(fasta, start=16, width=7))["CGGGCCT"]
table(subseq(fasta, start=17, width=7))["CGGGCCT"]
table(subseq(fasta, start=18, width=7))["CGGGCCT"]
table(subseq(fasta, start=19, width=7))["CGGGCCT"]
table(subseq(fasta, start=20, width=7))["CGGGCCT"]
table(subseq(fasta, start=21, width=7))["CGGGCCT"]
table(subseq(fasta, start=22, width=7))["CGGGCCT"]
table(subseq(fasta, start=23, width=7))["CGGGCCT"]
table(subseq(fasta, start=24, width=7))["CGGGCCT"]
table(subseq(fasta, start=25, width=7))["CGGGCCT"]
table(subseq(fasta, start=26, width=7))["CGGGCCT"]
table(subseq(fasta, start=27, width=7))["CGGGCCT"]
table(subseq(fasta, start=28, width=7))["CGGGCCT"]
table(subseq(fasta, start=29, width=7))["CGGGCCT"]
table(subseq(fasta, start=30, width=7))["CGGGCCT"]
table(subseq(fasta, start=31, width=7))["CGGGCCT"]
table(subseq(fasta, start=32, width=7))["CGGGCCT"]
table(subseq(fasta, start=33, width=7))["CGGGCCT"]
table(subseq(fasta, start=34, width=7))["CGGGCCT"]
table(subseq(fasta, start=35, width=7))["CGGGCCT"]
table(subseq(fasta, start=36, width=7))["CGGGCCT"]
table(subseq(fasta, start=37, width=7))["CGGGCCT"]
table(subseq(fasta, start=38, width=7))["CGGGCCT"]
table(subseq(fasta, start=39, width=7))["CGGGCCT"]
table(subseq(fasta, start=40, width=7))["CGGGCCT"]
table(subseq(fasta, start=41, width=7))["CGGGCCT"]
table(subseq(fasta, start=42, width=7))["CGGGCCT"]
table(subseq(fasta, start=43, width=7))["CGGGCCT"]
table(subseq(fasta, start=44, width=7))["CGGGCCT"]
table(subseq(fasta, start=45, width=7))["CGGGCCT"]
table(subseq(fasta, start=46, width=7))["CGGGCCT"]
table(subseq(fasta, start=47, width=7))["CGGGCCT"]
table(subseq(fasta, start=48, width=7))["CGGGCCT"]
table(subseq(fasta, start=49, width=7))["CGGGCCT"]
table(subseq(fasta, start=50, width=7))["CGGGCCT"]
table(subseq(fasta, start=51, width=7))["CGGGCCT"]
table(subseq(fasta, start=52, width=7))["CGGGCCT"]
table(subseq(fasta, start=53, width=7))["CGGGCCT"]
table(subseq(fasta, start=54, width=7))["CGGGCCT"]
table(subseq(fasta, start=55, width=7))["CGGGCCT"]
table(subseq(fasta, start=56, width=7))["CGGGCCT"]
table(subseq(fasta, start=57, width=7))["CGGGCCT"]
table(subseq(fasta, start=58, width=7))["CGGGCCT"]
table(subseq(fasta, start=59, width=7))["CGGGCCT"]
table(subseq(fasta, start=60, width=7))["CGGGCCT"]
table(subseq(fasta, start=61, width=7))["CGGGCCT"]
table(subseq(fasta, start=62, width=7))["CGGGCCT"]
table(subseq(fasta, start=63, width=7))["CGGGCCT"]
table(subseq(fasta, start=64, width=7))["CGGGCCT"]
table(subseq(fasta, start=65, width=7))["CGGGCCT"]
table(subseq(fasta, start=66, width=7))["CGGGCCT"]
table(subseq(fasta, start=67, width=7))["CGGGCCT"]
table(subseq(fasta, start=68, width=7))["CGGGCCT"]
table(subseq(fasta, start=69, width=7))["CGGGCCT"]
table(subseq(fasta, start=70, width=7))["CGGGCCT"]
table(subseq(fasta, start=71, width=7))["CGGGCCT"]
table(subseq(fasta, start=72, width=7))["CGGGCCT"]
table(subseq(fasta, start=73, width=7))["CGGGCCT"]
table(subseq(fasta, start=74, width=7))["CGGGCCT"]
table(subseq(fasta, start=75, width=7))["CGGGCCT"]
table(subseq(fasta, start=76, width=7))["CGGGCCT"]
table(subseq(fasta, start=77, width=7))["CGGGCCT"]
table(subseq(fasta, start=78, width=7))["CGGGCCT"]
table(subseq(fasta, start=79, width=7))["CGGGCCT"]
table(subseq(fasta, start=80, width=7))["CGGGCCT"]
table(subseq(fasta, start=81, width=7))["CGGGCCT"]
table(subseq(fasta, start=82, width=7))["CGGGCCT"]
table(subseq(fasta, start=83, width=7))["CGGGCCT"]
table(subseq(fasta, start=84, width=7))["CGGGCCT"]
table(subseq(fasta, start=85, width=7))["CGGGCCT"]
table(subseq(fasta, start=86, width=7))["CGGGCCT"]
table(subseq(fasta, start=87, width=7))["CGGGCCT"]
table(subseq(fasta, start=88, width=7))["CGGGCCT"]
table(subseq(fasta, start=89, width=7))["CGGGCCT"]
table(subseq(fasta, start=90, width=7))["CGGGCCT"]
table(subseq(fasta, start=91, width=7))["CGGGCCT"]
table(subseq(fasta, start=92, width=7))["CGGGCCT"]
table(subseq(fasta, start=93, width=7))["CGGGCCT"]
table(subseq(fasta, start=94, width=7))["CGGGCCT"]
table(subseq(fasta, start=95, width=7))["CGGGCCT"]
table(subseq(fasta, start=96, width=7))["CGGGCCT"]
table(subseq(fasta, start=97, width=7))["CGGGCCT"]
table(subseq(fasta, start=98, width=7))["CGGGCCT"]
table(subseq(fasta, start=99, width=7))["CGGGCCT"]
table(subseq(fasta, start=100, width=7))["CGGGCCT"]
table(subseq(fasta, start=101, width=7))["CGGGCCT"]
param_kmer <- 7
param_obj <- "CGGGCCT"
Obs <- NULL
for(i in 1:101){
Obs <- c(Obs, table(subseq(fasta, start=i, width=param_kmer))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
param_obj <- "CGGGCCT"
Obs <- NULL
for(i in 1:101){
Obs <- c(Obs, table(subseq(fasta, start=i, width=nchar(param_obj)))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
param_len_ngs <- 107
param_obj <- "CGGGCCT"
Obs <- NULL
hoge <- param_len_ngs - nchar(param_obj) + 1
for(i in 1:hoge){
Obs <- c(Obs, table(subseq(fasta, start=i, width=nchar(param_obj)))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
param_obj <- "CGGGCCT"
Obs <- NULL
hoge <- width(fasta)[1] - nchar(param_obj) + 1
for(i in 1:hoge){
Obs <- c(Obs, table(subseq(fasta, start=i, width=nchar(param_obj)))[param_obj])
}
Obs[is.na(Obs)] <- 0
head(Obs)
mean(Obs, na.rm=TRUE)
Exp <- mean(Obs, na.rm=TRUE)
head(Obs/Exp)
plot(Obs/Exp, type="l", col="red")
イントロ | NGS | 読み込み | FASTQ形式 | 応用
FASTQ形式ファイルを読み込むやり方を示します。ファイルサイズが数GBというレベルになってきてますので、
圧縮ファイルでの読み込みが基本となりつつあります。
しかし、一度に圧縮ファイル中の中身を一旦全て読み込むことも厳しい、あるいはもはやできない状態になってきています。
この背景を踏まえ、「応用」では、圧縮FASTQファイルを入力として、メモリオーバーフロー(スタックオーバーフロー;stack overflow)
にならないように一部だけを読み込む手順を示します。まだ途中段階ですが、野間口達洋 氏提供情報をもとにいくつか試しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. small RNA-seqのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られた
カイコsmall RNA-seqデータ(Nie et al., BMC Genomics, 2013)です。
入力ファイルサイズは400Mb弱、11,928,428リードです。この中から最初の100000リード分をgzip圧縮ファイルとして出力しています。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge1.fasta.gz"
param <- 100000
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq",
skip=0, nrec=param)
fasta
writeXStringSet(fasta, file=out_f,
format="fasta", compress=T, width=50)
2. small RNA-seqのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られた
2015年6月18日現在(R ver. 3.2.0; Biostrings ver. 2.36.1)、エラーを吐かずにFASTQ形式のgzip圧縮ファイルは得られるのですが、、、
クオリティスコア部分が変です。つまり、このやり方はダメです。
おそらくreadDNAStringSet関数で読み込んだ情報の中にクオリティスコアが含まれていないため、
ダミーの「;」というスコアがそのまま表示されているだけだと思います。
readDNAStringSet関数でクオリティスコアを読み込むやり方(あるいは取り出し方)が分かった方は教えて下さいm(_ _)m
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge2.fastq.gz"
param <- 100000
library(Biostrings)
fastq <- readDNAStringSet(in_f, format="fastq",
skip=0, nrec=param)
fastq
writeXStringSet(fastq, file=out_f,
format="fastq", compress=T)
3. small RNA-seqのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
原始的なやり方ですが、とりあえずうまく動きます。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge3.fastq"
param <- 100000
fastq <- readLines(in_f, n=param*4)
writeLines(fastq, con=out_f)
イントロ | NGS | 読み込み | FASTQ形式 | description行の記述を整形
FASTQ形式ファイルに対して、 「目的の記述部分のみ抽出し、それを新たなdescription」としてFASTQ形式で保存するやり方などを示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
抽出例:「SRR037439.1 HWI-E4_6_30ACL:2:1:0:176 length=35」-->「SRR037439.1」
戦略:description行の" "を区切り文字として分割("SRR037439.1"と"HWI-E4_6_30ACL:2:1:0:176"と"length=35")し、分割後の1つ目の要素を抽出
writeFastq関数実行時にcompress=Fとして非圧縮FASTQ形式ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge1.fastq"
param1 <- " "
param2 <- 1
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), param1, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param2))
fastq <- ShortReadQ(sread(fastq), quality(fastq), description)
id(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
抽出例:「SRR037439.1 HWI-E4_6_30ACL:2:1:0:176 length=35」-->「SRR037439.1」
戦略:description行の" "を区切り文字として分割("SRR037439.1"と"HWI-E4_6_30ACL:2:1:0:176"と"length=35")し、分割後の1つ目の要素を抽出
writeFastq関数実行時にcompress=Tとしてgzip圧縮FASTQ形式ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge2.fastq.gz"
param1 <- " "
param2 <- 1
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), param1, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param2))
fastq <- ShortReadQ(sread(fastq), quality(fastq), description)
id(fastq)
writeFastq(fastq, out_f, compress=T)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
抽出例:「SRR037439.1 HWI-E4_6_30ACL:2:1:0:176 length=35」-->「SRR037439.1」
戦略:description行の" "を区切り文字として分割("SRR037439.1"と"HWI-E4_6_30ACL:2:1:0:176"と"length=35")し、分割後の1つ目の要素を抽出
writeXStringSet関数実行時にcompress=Fとして非圧縮FASTA形式ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge3.fasta"
param1 <- " "
param2 <- 1
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), param1, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param2))
fasta <- sread(fastq)
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
抽出例:「SRR037439.1 HWI-E4_6_30ACL:2:1:0:176 length=35」-->「SRR037439.1」
戦略:description行の" "を区切り文字として分割("SRR037439.1"と"HWI-E4_6_30ACL:2:1:0:176"と"length=35")し、分割後の1つ目の要素を抽出
writeXStringSet関数実行時にcompress=Tとしてgzip圧縮FASTA形式ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge4.fasta.gz"
param1 <- " "
param2 <- 1
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), param1, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param2))
fasta <- sread(fastq)
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", compress=T, width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
抽出例:「SRR037439.1 HWI-E4_6_30ACL:2:1:0:176 length=35」-->「SRR037439.1」
戦略:description行の" "を区切り文字として分割("SRR037439.1"と"HWI-E4_6_30ACL:2:1:0:176"と"length=35")し、分割後の1つ目の要素を抽出。
次に、"."を区切り文字として分割("SRR037439"と"1")し、分割後の2つ目の要素を抽出
writeFastq関数実行時にcompress=Fとして非圧縮FASTQ形式ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge5.fastq"
param1 <- " "
param2 <- 1
param3 <- "."
param4 <- 2
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), param1, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param2))
hoge <- strsplit(as.character(description), param3, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param4))
fastq <- ShortReadQ(sread(fastq), quality(fastq), description)
id(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
抽出例:「SRR037439.1 HWI-E4_6_30ACL:2:1:0:176 length=35」-->「SRR037439.1」
戦略:description行の" "を区切り文字として分割("SRR037439.1"と"HWI-E4_6_30ACL:2:1:0:176"と"length=35")し、分割後の1つ目の要素を抽出。
次に、"."を区切り文字として分割("SRR037439"と"1")し、分割後の2つ目の要素を抽出
writeXStringSet関数実行時にcompress=Fとして非圧縮FASTA形式ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge6.fasta"
param1 <- " "
param2 <- 1
param3 <- "."
param4 <- 2
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
hoge <- strsplit(as.character(id(fastq)), param1, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param2))
hoge <- strsplit(as.character(description), param3, fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", param4))
fasta <- sread(fastq)
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", compress=F, width=50)
イントロ | NGS | 読み込み | Illuminaの*_seq.txt
Illuminaの出力ファイルで、初期のものは*_seq.txtというものらしいです。実際の生の配列データファイルは数百程度に分割されています。
ここでは、そのファイル群がおさめられているディレクトリ(例:tmp)を指定して、その中の「*_seq.txt」ファイル群について一気に行う方法を示します。
ここでは、(s_1_0001_seq.txtとs_1_0007_seq.txtの二つのファイルを含む;
それぞれ1394行および826行からなる)フォルダを指定して読み込み、multi-FASTA形式で出力するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 指定したディレクトリ中に*_seq.txtという形式のファイルしかないという前提のもとで行う場合:
out_f <- "hoge1.fasta"
param <- "kkk"
library(ShortRead)
colClasses <- c(rep(list(NULL), 4), "DNAString")
fasta <- readXStringColumns(getwd(), pattern="", colClasses = colClasses)[[1]]
names(fasta) <- paste(param, 1:length(fasta), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. param2で指定した"s_1_*_seq.txt"という感じのファイル名のもののみ読み込む場合:
out_f <- "hoge2.fasta"
param1 <- "kkk"
param2 <- "s_1_.*_seq.txt"
library(ShortRead)
files <- list.files(getwd(), pattern=param2)
strsplit(readLines(files[[1]],1), "\t")
colClasses <- c(rep(list(NULL), 4), "DNAString")
fasta <- readXStringColumns(getwd(), pattern=param2, colClasses=colClasses)[[1]]
names(fasta) <- paste(param1, 1:length(fasta), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | NGS | 読み込み | Illuminaの*_qseq.txt
Illuminaの出力ファイルで、比較的最近のは*_qseq.txtというものらしいです。実際の生の配列データファイルは数百程度に分割されています。ここでは、そのファイル群がおさめられているディレクトリ(例:tmp)を指定して、その中の「*_qseq.txt」ファイル群について一気に行う方法を示します。ここでは、(s_1_1_0005_qseq.txtとs_1_1_0015_qseq.txtの二つのファイルを含む; それぞれ114行および73行からなる)フォルダを指定して読み込み、multi-FASTA形式で出力するやり方を示します。
*_qseq.txtファイルは全部で11列あり、9列目が塩基配列情報、10列目がquality情報を含んでいるので、その列の部分のみを抽出しているだけです。尚、指定したディレクトリ中に*_qseq.txtという形式のファイルしかなく、全てのファイルを取り扱いたいという前提となっています。
FASTQ形式といっても様々なバリエーション(Illumina FASTQやSanger FASTQなど)があります。Illumina *_qseq.txt中のquality scoreの値をそのまま用いてFASTQ形式で保存すればIllumina FASTQ形式ファイル上で取り扱うスコアとなります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 配列情報のみ抽出したい場合:
param1 <- "kkk"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[9] <- c("DNAString")
fasta <- readXStringColumns(getwd(), pattern="", colClasses = colClasses)[[1]]
names(fasta) <- paste(param1, 1:length(fasta), sep="_")
fasta
2. Illumina FASTQ形式でdescription行が"kkk_..."のようにしたい場合:
param1 <- "kkk"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[9:10] <- c("DNAString", "BString")
hoge <- readXStringColumns(getwd(), pattern="", colClasses = colClasses)
description <- paste(param1, 1:length(hoge[[1]]), sep="_")
description <- BStringSet(description)
fastq <- ShortReadQ(hoge[[1]], hoge[[2]], description)
sread(fastq)
quality(fastq)
id(fastq)
3. Illumina FASTQ形式で、descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしたい場合:
つまり、descriptionのところをqseq形式ファイルの「3列目(レーン番号)」:「4列目」:「5列目」:「6列目」:「11列目(pass filterフラグ情報)」で表すということ。
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[c(3:6,9:11)] <- c("BString","BString","BString","BString","DNAString", "BString", "BString")
hoge <- readXStringColumns(getwd(), pattern="", colClasses=colClasses)
description <- paste(hoge[[1]], hoge[[2]], hoge[[3]], hoge[[4]], hoge[[7]], sep=":")
description <- BStringSet(description)
fastq <- ShortReadQ(hoge[[5]], hoge[[6]], description)
sread(fastq)
quality(fastq)
id(fastq)
イントロ | ファイル形式の変換 | BAM --> BED
BAM形式のマッピング結果ファイル(拡張子が"bam"という前提です)を読み込んでBED形式に変換するやり方を示します。拡張子が"bed"の出力ファイルが自動生成されます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample_RNAseq1.bam"
library(GenomicAlignments)
hoge <- readGAlignments(in_f)
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", in_f)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
in_f <- "sample_RNAseq1.bam"
library(Rsamtools)
param_what <- c("rname", "pos", "qwidth", "strand")
param_flag <- scanBamFlag(isUnmappedQuery=FALSE)
param_bam <- ScanBamParam(what=param_what, flag=param_flag)
hoge <- scanBam(in_f, param=param_bam)[[1]]
tmp <- cbind(as.character(hoge$rname),
hoge$pos,
(hoge$qwidth+hoge$pos-1))
out_f <- sub(".bam", ".bed", in_f)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
イントロ | ファイル形式の変換 | FASTQ --> FASTA
Sanger FASTQ形式ファイルを読み込んでFASTA形式で出力するやり方を示します。
gzip圧縮FASTQファイルを読み込んでgzip圧縮FASTAファイル出力することもできます(WindowsはできるがMacはできないらしい)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
配列長が同じであることが既知の場合です。
in_f <- "SRR037439.fastq"
out_f <- "hoge1.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
うまくいかない例です
in_f <- "sample2.fastq"
out_f <- "hoge2.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
うまくいく例です。
in_f <- "sample2.fastq"
out_f <- "hoge3.fasta"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
fasta <- sread(fastq)
names(fasta) <- id(fastq)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
配列長が同じであってもreadFastq関数で読み込むことができます。
in_f <- "SRR037439.fastq"
out_f <- "hoge4.fasta"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
fasta <- sread(fastq)
names(fasta) <- id(fastq)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
配列長が同じであることが既知の場合です。
gzip圧縮FASTQファイルを読み込んで非圧縮FASTAファイルで出力するやり方です。
in_f <- "SRR037439.fastq.gz"
out_f <- "hoge5.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
配列長が同じであることが既知の場合です。
gzip圧縮FASTQファイルを読み込んでgzip圧縮FASTAファイルで出力するやり方です。
in_f <- "SRR037439.fastq.gz"
out_f <- "hoge6.fasta.gz"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", compress=T, width=50)
イントロ | ファイル形式の変換 | Genbank --> FASTA
Genbank形式ファイルを読み込んでFASTA形式で出力するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
NM_138957のページからGenbank形式で保存したものですが、
デフォルトの拡張子が.gbではうまく保存できなかったので*.gbkと変更しています。
in_f <- "NM_138957.gbk"
out_f <- "hoge1.txt"
library(seqinr)
gb2fasta(source.file=in_f, destination.file=out_f)
イントロ | ファイル形式の変換 | GFF3 --> GTF
GFFはGeneral Feature Formatの略で、version 3とversion 2があります。
私の認識では、version 3がGFF3で、version 2がGTFです。しかしながら、GFF version 2を少し改良したものがGTF形式という意見もあるようです。
ここでは、rtracklayerパッケージを用いて、GFF3形式ファイルを読み込んでGTF形式(GFF version 2)で出力するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
出力ファイルはhoge1.gtfです。
in_f <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3"
out_f <- "hoge1.gtf"
library(rtracklayer)
export(import(in_f), out_f, format="gtf")
イントロ | ファイル形式の変換 | qseq --> FASTA
Illuminaの*_qseq.txtファイル群がおさめられている(例:s_1_1_0005_qseq.txtとs_1_1_0015_qseq.txtの二つのファイルを含む; それぞれ114行および73行からなる)フォルダを指定して読み込むやり方を示します。
*_qseq.txtファイルは全部で11列あり、9列目が塩基配列情報、10列目がquality情報を含んでいます。尚、指定したディレクトリ中に*_qseq.txtという形式のファイルしかなく、全てのファイルを取り扱いたいという前提となっています。また、descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしています。
FASTQ形式といっても様々なバリエーション(Illumina FASTQやSanger FASTQなど)があります。Illumina *_qseq.txt中のquality scoreの値をそのまま用いてFASTQ形式で保存すればIllumina FASTQ形式ファイルとなります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. description行が"kkk_..."のようにしたい場合:
out_f <- "hoge1.fasta"
param1 <- "kkk"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[9] <- c("DNAString")
fasta <- readXStringColumns(getwd(), pattern="", colClasses = colClasses)[[1]]
names(fasta) <- paste(param1, 1:length(fasta), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしたい場合:
つまり、descriptionのところをqseq形式ファイルの「3列目(レーン番号)」:「4列目」:「5列目」:「6列目」:「11列目(pass filterフラグ情報)」で表すということ。
out_f <- "hoge2.fasta"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[c(3:6,9:11)] <- c("BString","BString","BString","BString","DNAString", "BString", "BString")
hoge <- readXStringColumns(getwd(), pattern="", colClasses=colClasses)
description <- paste(hoge[[1]], hoge[[2]], hoge[[3]], hoge[[4]], hoge[[7]], sep=":")
description <- BStringSet(description)
fasta <- hoge[[5]]
names(fasta) <- description
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
イントロ | ファイル形式の変換 | qseq --> Illumina FASTQ
Illuminaの*_qseq.txtファイル群がおさめられている(例:s_1_1_0005_qseq.txtと
s_1_1_0015_qseq.txtの二つのファイルを含む; それぞれ114行および73行からなる)フォルダを指定して読み込むやり方を示します。
*_qseq.txtファイルは全部で11列あり、9列目が塩基配列情報、10列目がquality情報を含んでいます。
尚、指定したディレクトリ中に*_qseq.txtという形式のファイルしかなく、全てのファイルを取り扱いたいという前提となっています。
また、descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしています。
FASTQ形式といっても様々なバリエーション(Illumina FASTQやSanger FASTQなど)があります。
Illumina *_qseq.txt中のquality scoreの値をそのまま用いてFASTQ形式で保存すればIllumina FASTQ形式ファイルとなります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしたい場合:
descriptionのところをqseq形式ファイルの「3列目(レーン番号)」:「4列目」:「5列目」:「6列目」:「11列目(pass filterフラグ情報)」で表すやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
out_f <- "hoge1.fastq"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[c(3:6,9:11)] <- c("BString","BString","BString","BString","DNAString", "BString", "BString")
hoge <- readXStringColumns(getwd(), pattern="", colClasses=colClasses)
description <- paste(hoge[[1]], hoge[[2]], hoge[[3]], hoge[[4]], hoge[[7]], sep=":")
description <- BStringSet(description)
fastq <- ShortReadQ(hoge[[5]], hoge[[6]], description)
sread(fastq)
quality(fastq)
id(fastq)
writeFastq(fastq, out_f, compress=F)
2. descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしつつ、一番右側に"/2"の記述を追加したい場合:
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
out_f <- "hoge2.fastq"
param <- "/2"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[c(3:6,9:11)] <- c("BString","BString","BString","BString","DNAString", "BString", "BString")
hoge <- readXStringColumns(getwd(), pattern="", colClasses=colClasses)
description <- paste(hoge[[1]], hoge[[2]], hoge[[3]], hoge[[4]], hoge[[7]], sep=":")
description <- BStringSet(paste(description, param, sep=""))
fastq <- ShortReadQ(hoge[[5]], hoge[[6]], description)
sread(fastq)
quality(fastq)
id(fastq)
writeFastq(fastq, out_f, compress=F)
イントロ | ファイル形式の変換 | qseq --> Sanger FASTQ
Illuminaの*_qseq.txtファイル群がおさめられている(例:s_1_1_0005_qseq.txtと
s_1_1_0015_qseq.txtの二つのファイルを含む; それぞれ114行および73行からなる)フォルダを指定して読み込むやり方を示します。
*_qseq.txtファイルは全部で11列あり、9列目が塩基配列情報、10列目がquality情報を含んでいます。
尚、指定したディレクトリ中に*_qseq.txtという形式のファイルしかなく、全てのファイルを取り扱いたいという前提となっています。また、descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしています。
2013/08/18までは、例えば「Illumina ASCII 64」に相当する"@"を「Sanger ASCII 33」に相当する"!"などと一文字ずつ変換していましたが、forループを用いて一度に変換する美しいスクリプトに変更しました。
基本的にはIllumina ASCIIコードに「-31」することでSanger ASCIIコードに変換できるので、Phred score = 0に相当する「Illumina ASCII 64」から、Phred score = 61に相当する「Illumina ASCII 126」までの範囲をSanger ASCIIコードに変換しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしたい場合:
descriptionのところをqseq形式ファイルの「3列目(レーン番号)」:「4列目」:「5列目」:「6列目」:「11列目(pass filterフラグ情報)」で表すやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
out_f <- "hoge1.fastq"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[c(3:6,9:11)] <- c("BString","BString","BString","BString","DNAString", "BString", "BString")
hoge <- readXStringColumns(getwd(), pattern="", colClasses=colClasses)
description <- paste(hoge[[1]], hoge[[2]], hoge[[3]], hoge[[4]], hoge[[7]], sep=":")
description <- BStringSet(description)
qscore <- hoge[[6]]
for(i in 64:126){
qscore <- chartr(rawToChar(as.raw(i)), rawToChar(as.raw(i - 31)), qscore)
}
fastq <- ShortReadQ(hoge[[5]], qscore, description)
sread(fastq)
quality(fastq)
id(fastq)
writeFastq(fastq, out_f, compress=F)
2. descriptionのところが通常のqseq2fastq.plの出力結果とほぼ同等の形式にしつつ、一番右側に"/2"の記述を追加したい場合:
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
out_f <- "hoge2.fastq"
param <- "/2"
library(ShortRead)
colClasses <- rep(list(NULL), 11)
colClasses[c(3:6,9:11)] <- c("BString","BString","BString","BString","DNAString", "BString", "BString")
hoge <- readXStringColumns(getwd(), pattern="", colClasses=colClasses)
description <- paste(hoge[[1]], hoge[[2]], hoge[[3]], hoge[[4]], hoge[[7]], sep=":")
description <- BStringSet(paste(description, param, sep=""))
qscore <- hoge[[6]]
for(i in 64:126){
qscore <- chartr(rawToChar(as.raw(i)), rawToChar(as.raw(i - 31)), qscore)
}
fastq <- ShortReadQ(hoge[[5]], qscore, description)
sread(fastq)
quality(fastq)
id(fastq)
writeFastq(fastq, out_f, compress=F)
前処理 | クオリティコントロール | について
数億~数十億リードからなるNGSデータの全体的な精度チェック、クオリティの低いリードのフィルタリング、
リードに含まれるアダプター/プライマー配列(adapters/primers)やクオリティの低い配列部分の除去(トリミング; trimming)などを実行する様々な方法をリストアップします。
Krakenなどアダプター配列除去などが行えるものも含みます。
2014年ごろまでは、FASTQファイルに対するQuality Control (QC)用プログラムをリストアップしていましたが、
Lun et al., F1000Res., 2016のようなsingle-cell RNA-seq (scRNA-seq)のカウントデータのQCなども含めています。(2019年3月28日追加)。
R用:
- qrqc:原著論文なし
- fastqcr:原著論文なし
- PIQA: Martinez-Alcantara et al., Bioinformatics, 2009
- ShortRead:Morgan et al., Bioinformatics, 2009
- girafe:Toedling et al., Bioinformatics, 2010
- QuasR:Gaidatzis et al., Bioinformatics, 2015
- scater (scRNA-seq用):McCarthy et al., Bioinformatics, 2017
- yarn:Paulson et al., BMC Bioinformatics, 2017
R以外:
- FastQC:原著論文なし
- FASTX-Toolkit:原著論文なし
- SolexaQA:Cox et al., BMC Bioinformatics, 2010
- Quake:Kelley et al., Genome Biol., 2010
- NGSQC:Dai et al., BMC Genomics, 2010
- Cutadapt:Martin, M., EMBnet.journal, 2011
- PRINSEQ:Schmieder and Edwards, Bioinformatics, 2011
- ECHO:Kao et al., Genome Res., 2011
- Btrim:Kong Y., Genomics, 2011
- Hammer:Medvedev et al., Bioinformatics, 2011
- ConDeTri:Smeds et al., PLoS One, 2011
- BIGpre:Zhang et al., Genomics Proteomics Bioinformatics, 2011
- NGS QC Toolkit:Patel et al., PLoS One, 2012
- RobiNA:Lohse et al., Nucleic Acids Res., 2012
- SEQuel:Ronen et al., Bioinformatics, 2012
- AdapterRemoval:Lindgreen S., BMC Res Notes, 2012
- Slim-Filter:Golovko et al., BMC Bioinformatics, 2012
- HTQC:Yang et al., BMC Bioinformatics, 2013
- QC-Chain:Zhou et al., PLoS One, 2013
- Kraken:Davis et al., Methods, 2013
- AlienTrimmer:Criscuolo and Brisse, Genomics, 2013
- NextClip:Leggett et al., Bioinformatics, 2014
- QTrim (Roche/454などのlong read用):Shrestha et al., BMC Bioinformatics, 2014
- Trimmomatic:Bolger et al., Bioinformatics, 2014
- Skewer:Jiang et al., BMC Bioinformatics, 2014
- ngsShoRT:Chen et al., Source Code Biol Med., 2014
- FaQCs:Lo and Chain, BMC Bioinformatics, 2014
- UrQt:Modolo and Lerat, BMC Bioinformatics, 2015
- QoRTs:Hartley and Mullikin, BMC Bioinformatics, 2015
- SeqPurge (ngs-bitsの一部として提供されているようだ):Sturm et al., BMC Bioinformatics, 2016
- AfterQC:Chen et al., BMC Bioinformatics, 2017
- SOAPnuke:Chen et al., Gigascience, 2018
- RNA-QC-chain (QC-ChainのRNA-seq版):Zhou et al., BMC Genomics, 2018
- fastp:Chen et al., Bioinformatics, 2018
- FastqPuri:Pérez-Rubio et al., BMC Bioinformatics, 2019
- SequelTools(PacBio用):Hufnagel et al., BMC Bioinformatics, 2020
前処理 | クオリティチェック | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてQCレポートファイルを出力するやり方を示します。
FastQCのR版のようなものです。
qrqcよりも相当ストイックな出力結果です...。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
in_f <- "SRR037439.fastq.gz"
out_f <- "hoge1.pdf"
library(QuasR)
qQCReport(in_f, pdfFilename=out_f)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
in_f <- "SRR037439.fastq"
out_f <- "hoge2.pdf"
library(QuasR)
qQCReport(in_f, pdfFilename=out_f)
カイコsmall RNA-seqデータ(Nie et al., BMC Genomics, 2013; 約375MB)です。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge3.pdf"
library(QuasR)
qQCReport(in_f, pdfFilename=out_f)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約73MB)です。
in_f <- "SRR616268sub_1.fastq.gz"
out_f <- "hoge4.pdf"
library(QuasR)
qQCReport(in_f, pdfFilename=out_f)
前処理 | クオリティチェック | qrqc
FastQCのR版のようなものです。
Sanger FASTQ形式ファイルを読み込んで、positionごとの「クオリティスコア(quality score)」、
「どんな塩基が使われているのか(base frequency and base proportion)」、「リード長の分布」、
「GC含量」、「htmlレポート」などを出力してくれます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです
(Bullard et al., 2010)。
下記を実行すると「SRR037439-report」という名前のフォルダが作成されます。
中にあるreport.htmlをダブルクリックするとhtmlレポートを見ることができます。
in_f <- "SRR037439.fastq"
library(qrqc)
fastq <- readSeqFile(in_f, quality="sanger")
makeReport(fastq)
SRR633902から得られるFASTQファイルの最初の2,000行分を抽出したヒトデータです
(Chan et al., Hum. Mol. Genet., 2013)。
下記を実行すると「SRR633902_1_sub」という名前のフォルダが作成されます。中にあるreport.htmlをダブルクリックするとhtmlレポートを見ることができます。
「Error : Insufficient values in manual scale. 2 needed but only 1 provided.」というエラーが出ることは確認済みですが、htmlファイルは作成されます。
in_f <- "SRR633902_1_sub.fastq"
library(qrqc)
fastq <- readSeqFile(in_f, quality="sanger")
makeReport(fastq)
前処理 | フィルタリング | 任意のリード(サブセット)を抽出の8を実行して得られた
カイコsmall RNA-seqデータ(Nie et al., BMC Genomics, 2013; 100000リード; 約16MB)です。
下記を実行すると「SRR609266_sub」という名前のフォルダが作成されます。中にあるreport.htmlをダブルクリックするとhtmlレポートを見ることができます。
元のgzip圧縮FASTQファイルはNCBI SRAから取得したものであるため、本来はSanger Quality Scoreなはずです。
つまり、readSeqFile関数で読み込む際のデフォルトはquality="sanger"でよいはずです。
しかし、スコア分布的に用いたIllumina pipelineのバージョンが 1.3から1.7なんじゃないかなと思った(スコア分布が概ね30-40近辺にあるはずだという思想)ので、+33のSangerではなく+64のilluminaに独断で行っています。
このあたりの判定は現実問題としては、結構厄介です(Cock et al., 2010)。
in_f <- "SRR609266_sub.fastq"
library(qrqc)
fastq <- readSeqFile(in_f, quality="illumina")
makeReport(fastq)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
gzip圧縮ファイルの入力に対応していないのでエラーが出ます。
in_f <- "SRR037439.fastq.gz"
library(qrqc)
fastq <- readSeqFile(in_f, quality="sanger")
makeReport(fastq)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(gzip圧縮状態で約73MB)です。
qrqcパッケージはgzip圧縮ファイルの入力に対応していないので、自力でどうにかして解凍してください。
下記を実行すると「SRR609266_sub」という名前のフォルダが作成されます。中にあるreport.htmlをダブルクリックするとhtmlレポートを見ることができます。
元のgzip圧縮FASTQファイルはNCBI SRAから取得したものであるため、本来はSanger Quality Scoreなはずです。
つまり、readSeqFile関数で読み込む際のデフォルトはquality="sanger"でよいはずです。
しかし、スコア分布的に用いたIllumina pipelineのバージョンが 1.3から1.7なんじゃないかなと思った(スコア分布が概ね30-40近辺にあるはずだという思想)ので、+33のSangerではなく+64のilluminaに独断で行っています。
このあたりの判定は現実問題としては、結構厄介です(Cock et al., 2010)。
in_f <- "SRR616268sub_1.fastq"
library(qrqc)
fastq <- readSeqFile(in_f, quality="illumina")
makeReport(fastq)
前処理 | クオリティチェック | PHREDスコアに変換
ここでは、NCBI Sequence Read Archive (SRA)から得られる(従ってSanger FASTQ形式)FASTQ形式ファイルを読み込んでPHREDスコアに変換したり、そのboxplotを描画するやり方を示します。
PHREDスコアは、ベースコール(A, C, G, Tの四種類の塩基のうちのどの塩基かを決めること)のクオリティを示すp値(低ければ低いほどそのベースコールの信頼性が高いことを意味する)を二桁の整数値に変換したものであり、PHRED score = -10 * log10(p)で計算されます。
例えば、
- p=0.001の場合は、PHRED score = -10 * log10(0.001) = 30
- p=0.01の場合は、PHRED score = -10 * log10(0.01) = 20
- p=0.1の場合は、PHRED score = -10 * log10(0.1) = 10
- p=1の場合は、PHRED score = -10 * log10(1) = 0
のように計算されますので、PHREDスコアが高いほどそのベースコールの信頼性が高いことを意味します。
例題で解析しているファイルは全リードのほんの一部なので、ばらつきはありますが、5'側のベースコールのクオリティは高く、3'側に読み進んでいくほどPHREDスコアが低くなる傾向にあることが読み取れます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
PHREDスコア変換後、その数値情報をタブ区切りテキストファイルで保存するやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge1.txt"
library(ShortRead)
fastq <- readFastq(in_f)
out <- as(quality(fastq), "matrix")
dim(out)
colnames(out) <- 1:ncol(out)
rownames(out) <- as.character(id(fastq))
tmp <- cbind(rownames(out), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
PHREDスコア変換後、スコア分布をboxplotで描くやり方です。
in_f <- "SRR037439.fastq"
library(ShortRead)
fastq <- readFastq(in_f)
out <- as(quality(fastq), "matrix")
dim(out)
colnames(out) <- 1:ncol(out)
rownames(out) <- as.character(id(fastq))
boxplot(as.data.frame(out), outline=FALSE, xlab="cycle number", ylab="PHRED score")
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
PHREDスコア変換後、スコア分布のboxplotをpngファイルに保存するやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge3.png"
param_fig <- c(600, 400)
library(ShortRead)
fastq <- readFastq(in_f)
out <- as(quality(fastq), "matrix")
dim(out)
colnames(out) <- 1:ncol(out)
rownames(out) <- as.character(id(fastq))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
boxplot(as.data.frame(out), outline=FALSE, xlab="cycle number", ylab="PHRED score")
dev.off()
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
例題3と基本的に同じですが、y軸のPhredスコアの範囲を任意に指定するやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge4.png"
param_fig <- c(600, 400)
param_yrange <- c(5, 40)
library(ShortRead)
fastq <- readFastq(in_f)
out <- as(quality(fastq), "matrix")
dim(out)
colnames(out) <- 1:ncol(out)
rownames(out) <- as.character(id(fastq))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
boxplot(as.data.frame(out), outline=FALSE, xlab="cycle number", ylab="PHRED score", ylim=param_yrange)
dev.off()
前処理 | クオリティチェック | 配列長分布を調べる
FASTAまたはFASTQ形式ファイルを読み込んで配列長分布を得るやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample2.fasta"
out_f <- "hoge1.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- table(width(fasta))
out
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
in_f <- "sample2.fastq"
out_f <- "hoge2.txt"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
out <- table(width(fastq))
out
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
3. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(ファイルサイズは400Mb弱、11,928,428リード、配列長49 bp)です。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge3.txt"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
out <- table(width(fastq))
out
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
4. gzip圧縮FASTQ形式ファイル(hoge4.fastq.gz)の場合:
配列長の異なる仮想データファイルです。param_nbinsで50と指定すると、
1 bpおきに、1-50 bpまでのリード数が格納されます。
in_f <- "hoge4.fastq.gz"
out_f <- "hoge4.txt"
param_nbins <- 50
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
out <- tabulate(width(fastq), param_nbins)
out
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
5. gzip圧縮FASTQ形式ファイル(hoge4.fastq.gz)の場合:
配列長の異なる仮想データファイルです。param_nbinsで50と指定すると、
1 bpおきに、1-50 bpまでのリード数が格納されます。
QuasRパッケージ(Gaidatzis et al., 2015)
マニュアル中のbarplotを用いた作図形式に似せています。
in_f <- "hoge4.fastq.gz"
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5.png"
param_nbins <- 50
param_fig <- c(700, 400)
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
out <- tabulate(width(fastq), param_nbins)
out
names(out) <- 1:param_nbins
tmp <- cbind(names(out), out)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
barplot(out/sum(out)*100, xlab="Read length", ylab="Percent of reads")
grid(col="gray", lty="dotted")
dev.off()
前処理 | クオリティチェック | Overrepresented sequences | ShortRead(Morgan_2009)
ShortReadパッケージを用いてリードの種類ごとの出現回数をを得るやり方を示します。FastQCの
Overrepresented sequencesの項目と同じような結果が得られます。
前処理 | フィルタリング | 重複のない配列セットを作成も基本的にやっていることは同じです。
ファイルに保存部分で「tmp <- cbind(names(out), out)」と書くのは冗長であるとの指摘を受けたので修正しました(野間口達洋 氏提供情報; 2015/07/29追加)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(ファイルサイズは400Mb弱、11,928,428リード、配列長49 bp)です。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge1.txt"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
out <- table(sread(fastq))
out <- sort(out, decreasing=T)
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
2. gzip圧縮FASTQ形式ファイル(hoge4.fastq.gz)の場合:
配列長の異なる仮想データファイルです。
in_f <- "hoge4.fastq.gz"
out_f <- "hoge2.txt"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
out <- table(sread(fastq))
out <- sort(out, decreasing=T)
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約73MB)です。長さは全て107 bpです。
in_f <- "SRR616268sub_1.fastq.gz"
out_f <- "hoge3.txt"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
out <- table(sread(fastq))
out <- sort(out, decreasing=T)
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約73MB)です。長さは全て107 bpです。
3.と基本的に同じですが、FastQCのデフォルトと同じく、最初の50bp分のみで解析するやり方です。
「イントロ | 一般 | 指定した範囲の配列を取得」のテクニックとの併用になります。
in_f <- "SRR616268sub_1.fastq.gz"
out_f <- "hoge4.txt"
param <- c(1, 50)
library(ShortRead)
fastq <- readFastq(in_f)
fasta <- sread(fastq)
fasta
fasta <- subseq(fasta, start=param[1], end=param[2])
fasta
out <- table(fasta)
out <- sort(out, decreasing=T)
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=T, col.names=F)
前処理 | トリミング | ポリA配列除去 | ShortRead(Morgan_2009)
ShortReadパッケージを用いたmRNAの3'末端に存在するpoly A配列部分をトリムするやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
許容するミスマッチ数を0としています。
in_f <- "sample3.fasta"
out_f <- "hoge1.fasta"
param1 <- "AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA"
param_mismatch <- 0
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- trimLRPatterns(Rpattern=param1, subject=fasta, max.Rmismatch=rep(param_mismatch, nchar(param1)))
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
許容するミスマッチ数を1としています。
in_f <- "sample3.fasta"
out_f <- "hoge2.fasta"
param1 <- "AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA"
param_mismatch <- 1
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- trimLRPatterns(Rpattern=param1, subject=fasta, max.Rmismatch=rep(param_mismatch, nchar(param1)))
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
前処理 | トリミング | アダプター配列除去(基礎) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いたアダプター/プライマー配列除去の基本形を示します。
param_nrecオプションは、一度に処理するリード数を指定しているところですが、基本的に無視で構いません。
デフォルトの1000000のときに、メモリ不足でフリーズしたので、デフォルトの半分の500000リードにしています。
処理時間はおそらく長くなりますが、エラーなく動くことのほうが重要です。
それでもフリーズするヒトは、ここの数値を200000などさらに小さくして対処してください。
2015年6月24日にparam_nrecが適切に反映されるように修正しました(中村浩正 氏提供情報)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(ファイルサイズは400Mb弱、11928428リード)です。
原著論文(Nie et al., BMC Genomics, 2013)中の記述から
GSE41841を頼りに、
SRP016842にたどりつき、
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものが入力ファイルです。
原著論文の情報から、おそらくアダプター配列は"TGGAATTCTCGGGTGCCAAGGAACTCCAGTC..."という感じだろうと推測できます。
ここでは、アダプター配列以外はデフォルトで実行しています。
アダプター配列の位置は5'側(左側)ではなく3'側(右側)にあるという前提であり、右側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にRpatternのみ記載している理由です。約5分。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge1.fastq.gz"
param_adapter <- "TGGAATTCTCGGGTGCCAAGGAACTCCAGTC"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Rpattern=param_adapter,
nrec=param_nrec)
res
2. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(ファイルサイズは400Mb弱、11928428リード)です。
このファイルに対するFastQC実行結果として
「RNA PCR Primer, Index 1」(RPI1)が含まれているとレポートされました。これでググると塩基配列情報は
"CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA"
と書いてあったので、これを入力として与えます。
アダプター配列の位置は5'側(左側)ではなく3'側(右側)にあるという前提であり、右側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にRpatternのみ記載している理由です。約5分。
このやり方では、結論としてはうまくいきません。
理由はおそらくparam_adapterで指定している配列はプライマー配列なので、これの逆相補鎖(reverse complement)を与えないといけないんだと思います。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge2.fastq.gz"
param_adapter <- "CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Rpattern=param_adapter,
nrec=param_nrec)
res
3. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
2.の対策としてparam_adapterとして与える配列は同じで、内部的に逆相補鎖(reverse complement)を作成して与えるやり方です。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge3.fastq.gz"
param_adapter <- "CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA"
param_nrec <- 500000
library(QuasR)
library(Biostrings)
hoge <- reverseComplement(DNAString(param_adapter))
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Rpattern=as.character(hoge),
nrec=param_nrec)
res
4. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(ファイルサイズは400Mb弱、11928428リード)です。
このファイルに対するFastQC実行結果として
「RNA PCR Primer, Index 1」(RPI1)が含まれているとレポートされた49 bpをアダプター配列として入力しています。
ここでは、アダプター配列以外はデフォルトで実行しています。
アダプター配列の位置は5'側(左側)ではなく3'側(右側)にあるという前提であり、右側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にRpatternのみ記載している理由です。約5分。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge4.fastq.gz"
param_adapter <- "TGGAATTCTCGGGTGCCAAGGAACTCCAGTCACATCACGATCTCGTATG"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Rpattern=param_adapter,
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分です。paired-endデータのforward側、約75MB(74,906,576 bytes)、全リード107 bpです。
FastQC実行結果(SRR616268sub_1_fastqc.html)として
「TruSeq Adapter, Index 3」が含まれているとレポートされました。これでググると塩基配列情報は
"GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG"
と書いてあったので、これを入力として与えます。
アダプター配列の位置は5'側(左側)にあるという前提であり、左側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にLpatternのみ記載している理由です。約2分。
出力ファイル(998,664リード、71,426,499 bytes)でもう一度FastQCを実行すると、「TruSeq Adapter, Index 3」がOverrepresented sequencesの項目から消えていることまでは確認しました。
しかし、まだ「TruSeq Adapter, Index 2」が残っています。
許容するN数(nBases)は1、最短配列長(minLength)は18がデフォルトなのでそれが自動的に適用されます。
in_f <- "SRR616268sub_1.fastq.gz"
out_f <- "hoge5.fastq.gz"
param_adapter <- "GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Lpattern=param_adapter,
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
このファイルに対するFastQC実行結果
(Overrepresented sequencesの項目)中には「TruSeq Adapter, Index 3」と「TruSeq Adapter, Index 2」が含まれているとレポートされました。
TruSeq Adapter, Index 1-27の塩基配列は、任意の一文字を表す「.」を用いることで
"GATCGGAAGAGCACACGTCTGAACTCCAGTCAC......ATCTCGTATGCCGTCTTCTGCTTG"
とすることができますので、これを入力として与えます。約2分。
現状ではエラーは吐きませんが、うまくトリムできないようです。正規表現に対応していないか、
ミスマッチとしてカウントされてしまっているか、、、でしょうか。
in_f <- "SRR616268sub_1.fastq.gz"
out_f <- "hoge6.fastq.gz"
param_adapter <- "GATCGGAAGAGCACACGTCTGAACTCCAGTCAC......ATCTCGTATGCCGTCTTCTGCTTG"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Lpattern=param_adapter,
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
7. gzip圧縮FASTQ形式ファイル(hoge5.fastq.gz)の場合:
5.で得られたhoge5.fastq.gzファイル中には、
まだ「TruSeq Adapter, Index 2」が残っています。
「TruSeq Adapter, Index 2」の塩基配列情報は
"GATCGGAAGAGCACACGTCTGAACTCCAGTCACCGATGTATCTCGTATGCCGTCTTCTGCTTG"
と書いてあったので、これを入力として与えます。約2分。
この出力ファイル(hoge7.fastq.gz)と同じものが
SRR616268sub_trim_1.fastq.gz(998,658リード、71,227,695 bytes)です。
FastQC実行結果はSRR616268sub_trim_1_fastqc.htmlです。
許容するN数(nBases)は1、最短配列長(minLength)は18がデフォルトなのでそれが自動的に適用されます。
in_f <- "hoge5.fastq.gz"
out_f <- "hoge7.fastq.gz"
param_adapter <- "GATCGGAAGAGCACACGTCTGAACTCCAGTCACCGATGTATCTCGTATGCCGTCTTCTGCTTG"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Lpattern=param_adapter,
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分です。paired-endデータのreverse側、約67MB、全リード93 bpです。
FastQC実行結果(SRR616268sub_2_fastqc.html)として
「Illumina Single End PCR Primer 1」が含まれているとレポートされました。これでググると塩基配列情報は
"AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT"
と書いてあったので、これを入力として与えます。
アダプター配列の位置は5'側(左側)にあるという前提であり、左側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にLpatternのみ記載している理由です。約2分。
この出力ファイル(hoge8.fastq.gz)と同じものが
SRR616268sub_trim_2.fastq.gz(999,136リード、63,442,904 bytes)です。
FastQC実行結果はSRR616268sub_trim_2_fastqc.htmlです。
許容するN数(nBases)は1、最短配列長(minLength)は18がデフォルトなのでそれが自動的に適用されます。
in_f <- "SRR616268sub_2.fastq.gz"
out_f <- "hoge8.fastq.gz"
param_adapter <- "AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT"
param_nrec <- 500000
library(QuasR)
library(Biostrings)
hoge <- reverseComplement(DNAString(param_adapter))
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Lpattern=as.character(hoge),
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
前処理 | トリミング | アダプター配列除去(基礎) | girafe(Toedling_2010)
girafeパッケージを用いたアダプター配列除去を行うやり方を示します。アダプター配列除去を行うやり方を示します。
注意点1としては、実際に塩基配列長が短くなっていてもdescription行の記述(特に配列長情報の記述)は変わりませんので、「なんかおかしい」と気にしなくて大丈夫です。
注意点2としては、例えば、アダプター配列の5'側が「CATCG...」となっているにも関わらずなぜ二番目の配列の3'側「...CATAG」の最後の5塩基がトリムされているのだろう?と疑問に思われる方がいらっしゃるかもしれませんが、これは。R Console上で「?trimAdapter」と打ち込んでデフォルトのオプションを眺めることで理由がわかります。つまり、アラインメントスコア計算時に、この関数はデフォルトで一致に1点、不一致に-1点を与えて一塩基ずつオーバーラップの度合いを上げていく、という操作をしているからです。
したがって、もし完全一致のみに限定したい場合は、trimAdapter関数のところで、不一致に対して大幅に減点するようなパラメータを与えればいいんです。例えば「mismatch.score = -1000」とか。。。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
デフォルトのパラメータ(一致に1点、不一致に-1点)でトリムし、FASTQ形式で保存するやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge1.fastq"
param_adapter <- "CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG"
library(girafe)
fastq <- readFastq(in_f)
sread(fastq)
table(width(fastq))
fastq <- trimAdapter(fastq, param_adapter)
sread(fastq)
table(width(fastq))
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
アダプター配列と完全一致のみで2塩基以上一致するものだけをトリムし、FASTQ形式で保存するやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge2.fastq"
param_adapter <- "CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG"
param2 <- 2
library(girafe)
fastq <- readFastq(in_f)
sread(fastq)
table(width(fastq))
mismatch <- nchar(param_adapter) + 100
fastq <- trimAdapter(fastq, param_adapter,
mismatch.score = -mismatch,
score.threshold = param2)
sread(fastq)
table(width(fastq))
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
アダプター配列と完全一致のみで2塩基以上一致するものだけをトリムし、FASTA形式で保存するやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge3.fasta"
param_adapter <- "CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG"
param2 <- 2
library(girafe)
fastq <- readFastq(in_f)
sread(fastq)
table(width(fastq))
mismatch <- nchar(param_adapter) + 100
fastq <- trimAdapter(fastq, param_adapter,
mismatch.score = -mismatch,
score.threshold = param2)
fasta <- sread(fastq)
names(fasta) <- id(fastq)
fasta
table(width(fasta))
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
アダプター配列と完全一致のみで2塩基以上一致するものだけをトリムし、gzip圧縮FASTQ形式ファイルで保存するやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
in_f <- "SRR037439.fastq"
out_f <- "hoge4.fastq.gz"
param_adapter <- "CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG"
param2 <- 2
library(girafe)
fastq <- readFastq(in_f)
sread(fastq)
table(width(fastq))
mismatch <- nchar(param_adapter) + 100
fastq <- trimAdapter(fastq, param_adapter,
mismatch.score = -mismatch,
score.threshold = param2)
sread(fastq)
table(width(fastq))
writeFastq(fastq, out_f, compress=T)
前処理 | トリミング | アダプター配列除去(基礎) | ShortRead(Morgan_2009)
ShortReadパッケージを用いたアダプター配列除去を行うやり方を示します。
smallRNA-seqから得られる配列データにはアダプター配列(例:CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG; Oligonucleotide sequences (c) 2007-2009 Illumina, Inc. All rights reserved.)
が含まれ、アダプター配列を含んだままの状態でゲノムなどのリファレンス配列にマッピングすることはできません。
ここでは、許容するミスマッチ数を指定して、RNA-seqデータの右側にあるアダプター配列を除去するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
アダプター配列既知("ACGTACGTAA")で、許容するミスマッチ数が0 (param_mismatch <- 0)、
でアダプター配列除去を行い、FASTA形式で保存するやり方です。
in_f <- "sample3.fasta"
out_f <- "hoge1.fasta"
param_adapter <- "ACGTACGTAA"
param_mismatch <- 0
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- trimLRPatterns(Rpattern=param_adapter,
subject=fasta,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
アダプター配列既知("CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG")で、
許容するミスマッチ数が2 (param_mismatch <- 2)でアダプター配列除去を行い、FASTA形式で保存するやり方です。
in_f <- "sample1.fastq"
out_f <- "hoge2.fasta"
param_adapter <- "CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG"
param_mismatch <- 2
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
fasta <- trimLRPatterns(Rpattern=param_adapter,
subject=fasta,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
アダプター配列既知("CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG")で、
許容するミスマッチ数が2 (param_mismatch <- 2)でアダプター配列除去を行い、FASTQ形式で保存するやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "sample1.fastq"
out_f <- "hoge3.fastq"
param_adapter <- "CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG"
param_mismatch <- 2
library(ShortRead)
fastq <- readFastq(in_f)
hoge1 <- trimLRPatterns(Rpattern=param_adapter,
subject=sread(fastq),
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
hoge2 <- BStringSet(quality(quality(fastq)), start=1, end=width(hoge1))
fastq <- ShortReadQ(hoge1, hoge2, id(fastq))
sread(fastq)
quality(fastq)
writeFastq(fastq, out_f, compress=F)
4. FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(400Mb弱、11928428リード)です。
原著論文(Nie et al., BMC Genomics, 2013)中の記述から
GSE41841を頼りに、
SRP016842にたどりつき、
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものが入力ファイルです。
原著論文中では、アダプター配列やクオリティの低いリードを除去したのち、ゲノムにマッピングしたと書いてあります。
アダプター配列情報はどこにも書かれていませんでしたが、Table S2中のアダプター配列除去後の最も短いリードが18 nt (例:"GCAGTCGTGGCCGAGCGG")であり、
「この18 nt」と「この配列を含む生リード配列の差分」がアダプター配列ということになります。
詳細な情報は書かれていませんでしたが、おそらくアダプター配列は"TGGAATTCTCGGGTGCCAAGGAACTCCAGTC..."という感じだろうと推測して、
許容するミスマッチ数が1という条件でアダプタ配列除去を行っています。
最後のwriteFastq関数実行時にcompress=Tとしてgzip圧縮FASTQ形式で保存しています。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge4.fastq.gz"
param_adapter <- "TGGAATTCTCGGGTGCCAAGGAACTCCAGTC"
param_mismatch <- 1
library(ShortRead)
fastq <- readFastq(in_f)
hoge1 <- trimLRPatterns(Rpattern=param_adapter,
subject=sread(fastq),
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
hoge2 <- BStringSet(quality(quality(fastq)), start=1, end=width(hoge1))
fastq <- ShortReadQ(hoge1, hoge2, id(fastq))
sread(fastq)
quality(fastq)
writeFastq(fastq, out_f, compress=T)
前処理 | トリミング | アダプター配列除去(応用) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いたアダプター/プライマー配列除去とそれに付随する様々な組み合わせのやり方を示します。
エラーが発生してR Guiが終了することがありますが、この原因はメモリ不足だそうです(孫建強 氏提供情報)。
それゆえ、2015年6月21日にparam_nrecを追加しました。これは一度に処理するリード数を指定しているところですが、基本的に無視で構いません。。
デフォルトの1000000のときに、メモリ不足でフリーズしたので、デフォルトの半分のリード数にしています。
処理時間は長くなりますが、エラーなく動くことのほうが重要です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(ファイルサイズは400Mb弱、11928428リード)です。
原著論文(Nie et al., BMC Genomics, 2013)中の記述から
GSE41841を頼りに、
SRP016842にたどりつき、
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものが入力ファイルです。
原著論文中では、アダプター配列やクオリティの低いリードを除去したのち、ゲノムにマッピングしたと書いてあります。
アダプター配列情報はどこにも書かれていませんでしたが、Table S2中のアダプター配列除去後の最も短いリードが18 nt (例:"GCAGTCGTGGCCGAGCGG")であり、
「この18 nt」と「この配列を含む生リード配列の差分」がアダプター配列ということになります。
詳細な情報は書かれていませんでしたが、おそらくアダプター配列は"TGGAATTCTCGGGTGCCAAGGAACTCCAGTC..."という感じだろうと推測できます。
ここでは、1塩基ミスマッチまで許容して(推定)アダプター配列除去を行ったのち、
"ACGT"のみからなる配列(許容するN数が0)で、
配列長が18nt以上のものをフィルタリングして出力しています。
アダプター配列の位置は5'側(左側)ではなく3'側(右側)にあるという前提であり、右側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にRpatternとmax.Rmismatchのみ記載している理由です。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge1.fastq.gz"
param_adapter <- "TGGAATTCTCGGGTGCCAAGGAACTCCAGTC"
param_mismatch <- 1
param_nBases <- 0
param_minLength <- 18
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Rpattern=param_adapter,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)),
nBases=param_nBases,
minLength=param_minLength,
nrec=param_nrec)
res
乳酸菌RNA-seqデータSRR616268の最初の100万リード分です。paired-endデータです。
SRR616268sub_1.fastq.gzは、約75MB、全リード107 bpです。
SRR616268sub_2.fastq.gzは、約67MB、全リード93 bpです。
FastQC実行結果として
「TruSeq Adapter, Index 3」が含まれているとレポートされました。これでググると塩基配列情報は
"GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG"
と書いてあったので、これを入力として与えます。
アダプター配列の位置は5'側(左側)にあるという前提であり、左側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にLpatternのみ記載している理由です。
残念ながら、QuasR (ver. 1.8.2)では「Removing adapters from paired-end samples is not yet supported」と出ます。
2015年6月23日に開発者に実装を心待ちにしているとメールをしたら、翌日に対処法を指南していただきました(Thanks to Dr. Stadler)。
in_f1 <- "SRR616268sub_1.fastq.gz"
in_f2 <- "SRR616268sub_2.fastq.gz"
out_f1 <- "hoge1_1.fastq.gz"
out_f2 <- "hoge1_2.fastq.gz"
param_adapter <- "GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f1,
filenameMate=in_f2,
outputFilename=out_f1,
outputFilenameMate=out_f2,
Lpattern=param_adapter,
nrec=param_nrec)
res
乳酸菌RNA-seqデータSRR616268の最初の100万リード分です。paired-endデータのforward側、約75MB、全リード107 bpです。
FastQC実行結果(SRR616268sub_1_fastqc.html)として
「TruSeq Adapter, Index 3」が含まれているとレポートされました。これでググると塩基配列情報は
"GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG"
と書いてあったので、これを入力として与えます。
アダプター配列の位置は5'側(左側)にあるという前提であり、左側のアダプター配列しかトリムしないやり方です。
この例題では、アダプター配列除去のみしか行わず出力ファイル中のリード数が入力ファイルと同じになるようにしています。
これを確実に実現するために、許容するNの数としてトテツモナイ大きさの1000をparam_nBasesで与え、
アダプター配列除去後の許容する最短配列長として全てが残る0をparam_minLengthで与えています。
それが、preprocessReads関数実行時にLpatternのみ記載している理由です。約1分。
出力ファイルでもう一度FastQCを実行すると、「TruSeq Adapter, Index 3」がOverrepresented sequencesの項目から消えていることまでは確認しました。
しかし、まだ「TruSeq Adapter, Index 2」が残っています。
ファイルサイズが、74,906,576 bytesから71,542,907 bytesに若干減っていることがわかります。
in_f <- "SRR616268sub_1.fastq.gz"
out_f <- "hoge3.fastq.gz"
param_adapter <- "GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG"
param_nBases <- 1000
param_minLength <- 0
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Lpattern=param_adapter,
nBases=param_nBases,
minLength=param_minLength,
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
4. gzip圧縮FASTQ形式ファイル(hoge3.fastq.gz)の場合:
3.で得られたhoge3.fastq.gzファイル中には、
まだ「TruSeq Adapter, Index 2」が残っています。
「TruSeq Adapter, Index 2」の塩基配列情報は
"GATCGGAAGAGCACACGTCTGAACTCCAGTCACCGATGTATCTCGTATGCCGTCTTCTGCTTG"
と書いてあったので、これを入力として与えます。約1分。
ファイルサイズが、71,542,907 bytesから71,343,605 bytesに若干減っていることがわかります。
この出力ファイル(hoge4.fastq.gz)と同じものが
SRR616268sub_trim2_1.fastq.gz(1,000,000リード、71,343,605 bytes)です。
FastQC実行結果はSRR616268sub_trim2_1_fastqc.htmlです。
in_f <- "hoge3.fastq.gz"
out_f <- "hoge4.fastq.gz"
param_adapter <- "GATCGGAAGAGCACACGTCTGAACTCCAGTCACCGATGTATCTCGTATGCCGTCTTCTGCTTG"
param_nBases <- 1000
param_minLength <- 0
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Lpattern=param_adapter,
nBases=param_nBases,
minLength=param_minLength,
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分です。paired-endデータのreverse側、約67MB、全リード93 bpです。
FastQC実行結果(SRR616268sub_2_fastqc.html)として
「Illumina Single End PCR Primer 1」が含まれているとレポートされました。これでググると塩基配列情報は
"AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT"
と書いてあったので、これを入力として与えます。
アダプター配列の位置は5'側(左側)にあるという前提であり、左側のアダプター配列しかトリムしないやり方です。
それが、preprocessReads関数実行時にLpatternのみ記載している理由です。約1分。
ファイルサイズが、67,158,462 bytesから63,524,791 bytesに若干減っていることがわかります。
この出力ファイル(hoge5.fastq.gz)と同じものが
SRR616268sub_trim2_2.fastq.gz(1,000,000リード、63,524,791 bytes)です。
FastQC実行結果はSRR616268sub_trim2_2_fastqc.htmlです。
in_f <- "SRR616268sub_2.fastq.gz"
out_f <- "hoge5.fastq.gz"
param_adapter <- "AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT"
param_nBases <- 1000
param_minLength <- 0
param_nrec <- 500000
library(QuasR)
library(Biostrings)
hoge <- reverseComplement(DNAString(param_adapter))
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Lpattern=as.character(hoge),
nBases=param_nBases,
minLength=param_minLength,
nrec=param_nrec)
res
file.size(in_f)
file.size(out_f)
前処理 | トリミング | アダプター配列除去(応用) | ShortRead(Morgan_2009)
ShortReadパッケージを用いたアダプター配列除去とそれに付随する様々な組み合わせのやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
アダプター配列既知("ACGTACGTAA")で、許容するミスマッチ数が0 (param_mismatch <- 0)、
ACGTのみからなる配列(param_nBases <- 0)、配列長の範囲指定(20:30)
の組み合わせです。
in_f <- "sample3.fasta"
out_f <- "hoge1.fasta"
param_adapter <- "ACGTACGTAA"
param_mismatch <- 0
param_nBases <- 0
param_range <- 20:30
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- trimLRPatterns(Rpattern=param_adapter,
subject=fasta,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param_nBases
fasta <- fasta[obj]
fasta
obj <- (width(fasta) >= min(param_range)) & (width(fasta) <= max(param_range))
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
アダプター配列既知("CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG")で、許容するミスマッチ数が2 (param_mismatch <- 2)、
ACGTのみからなる配列(param_nBases <- 0)、配列長の範囲指定(20:30)
の組み合わせです。
in_f <- "sample1.fastq"
out_f <- "hoge2.fasta"
param_adapter <- "CATCGATCCTGCAGGCTAGAGACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG"
param_mismatch <- 2
param_nBases <- 0
param_range <- 20:30
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
fasta <- trimLRPatterns(Rpattern=param_adapter,
subject=fasta,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param_nBases
fasta <- fasta[obj]
fasta
obj <- (width(fasta) >= min(param_range)) & (width(fasta) <= max(param_range))
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. FASTQ形式ファイル(SRR609266.fastq)の場合:
small RNA-seqデータ(ファイルサイズは1.8GB、圧縮後で400Mb弱、11928428リード)です。
原著論文(Nie et al., BMC Genomics, 2013)中の記述から
GSE41841を頼りに、
SRP016842にたどりつき、
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものが入力ファイルです。
原著論文中では、アダプター配列やクオリティの低いリードを除去したのち、ゲノムにマッピングしたと書いてあります。
アダプター配列情報はどこにも書かれていませんでしたが、Table S2中のアダプター配列除去後の最も短いリードが18 nt (例:"GCAGTCGTGGCCGAGCGG")であり、
「この18 nt」と「この配列を含む生リード配列の差分」がアダプター配列ということになります。
詳細な情報は書かれていませんでしたが、おそらくアダプター配列は"TGGAATTCTCGGGTGCCAAGGAACTCCAGTC..."という感じだろうと推測できます。
アダプター配列既知("TGGAATTCTCGGGTGCCAAGGAACTCCAGTC")で、許容するミスマッチ数が2、
ACGTのみからなる配列(param_nBases <- 0)、配列長の範囲指定(20:30)
の組み合わせです。
in_f <- "SRR609266.fastq"
out_f <- "hoge3.fasta"
param_adapter <- "TGGAATTCTCGGGTGCCAAGGAACTCCAGTC"
param_mismatch <- 2
param_nBases <- 0
param_range <- 20:30
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
fasta <- trimLRPatterns(Rpattern=param_adapter,
subject=fasta,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param_nBases
fasta <- fasta[obj]
fasta
obj <- (width(fasta) >= min(param_range)) & (width(fasta) <= max(param_range))
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(400Mb弱、11928428リード)です。圧縮ファイルもreadDNAStringSet関数で通常手順で読み込めます。
原著論文(Nie et al., BMC Genomics, 2013)中の記述から
GSE41841を頼りに、
SRP016842にたどりつき、
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものが入力ファイルです。
原著論文中では、アダプター配列やクオリティの低いリードを除去したのち、ゲノムにマッピングしたと書いてあります。
アダプター配列情報はどこにも書かれていませんでしたが、Table S2中のアダプター配列除去後の最も短いリードが18 nt (例:"GCAGTCGTGGCCGAGCGG")であり、
「この18 nt」と「この配列を含む生リード配列の差分」がアダプター配列ということになります。
詳細な情報は書かれていませんでしたが、おそらくアダプター配列は"TGGAATTCTCGGGTGCCAAGGAACTCCAGTC..."という感じだろうと推測できます。
アダプター配列既知("TGGAATTCTCGGGTGCCAAGGAACTCCAGTC")で、許容するミスマッチ数が2、
ACGTのみからなる配列(param_nBases <- 0)、配列長の範囲指定(20:30)
の組み合わせです。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge4.fasta.gz"
param_adapter <- "TGGAATTCTCGGGTGCCAAGGAACTCCAGTC"
param_mismatch <- 2
param_nBases <- 0
param_range <- 20:30
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
fasta <- trimLRPatterns(Rpattern=param_adapter,
subject=fasta,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)))
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param_nBases
fasta <- fasta[obj]
fasta
obj <- (width(fasta) >= min(param_range)) & (width(fasta) <= max(param_range))
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50, compress=T)
前処理 | トリミング | 指定した末端塩基数だけ除去
3'末端を指定塩基数分だけトリムするやり方を示します。
トリム後の出力ファイルでdescription行部分の記述は不変ですのでご注意ください。理由は、この部分がただの文字列だからです。
2017.11.08の変更はこの部分の注意書きのみであり、コード自体はどこもいじってません。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. multi-FASTAファイル(hoge4.fa)の場合:
イントロ | 一般 | ランダムな塩基配列を作成の4.を実行して得られたファイルです。
in_f <- "hoge4.fa"
out_f <- "hoge1.fasta"
param_trim <- 5
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- width(fasta) - param_trim
hoge[hoge < 1] <- 1
fasta <- DNAStringSet(fasta, start=1, end=hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです
(Bullard et al., 2010)。
FASTQ形式ファイルを読み込んでFASTQ形式で保存するやり方です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge2.fastq"
param_trim <- 2
library(Biostrings)
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- width(sread(fastq)) - param_trim
hoge[hoge < 1] <- 1
hoge1 <- DNAStringSet(sread(fastq), start=1, end=hoge)
hoge2 <- BStringSet(quality(quality(fastq)), start=1, end=hoge)
fastq <- ShortReadQ(hoge1, hoge2, id(fastq))
sread(fastq)
quality(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです
(Bullard et al., 2010)。FASTQ形式ファイルを読み込んでFASTA形式で保存するやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge3.fasta"
param_trim <- 4
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
hoge <- width(fasta) - param_trim
hoge[hoge < 1] <- 1
fasta <- DNAStringSet(fasta, start=1, end=hoge)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約73MB; 74,906,576 bytes)です。
paired-endのforward側です。長さは全て107 bpです。
3'側の7 bp分をトリムするので、出力ファイル中の長さは100 bpになります。
in_f <- "SRR616268sub_1.fastq.gz"
out_f <- "hoge4.fastq.gz"
param_trim <- 7
library(Biostrings)
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- width(sread(fastq)) - param_trim
hoge[hoge < 1] <- 1
hoge1 <- DNAStringSet(sread(fastq), start=1, end=hoge)
hoge2 <- BStringSet(quality(quality(fastq)), start=1, end=hoge)
fastq <- ShortReadQ(hoge1, hoge2, id(fastq))
sread(fastq)
quality(fastq)
writeFastq(fastq, out_f, compress=T)
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約67MB; 67,158,462 bytes)です。
paired-endのforward側です。長さは全て93 bpです。
3'側の2 bp分をトリムするので、出力ファイル中の長さは91 bpになります。
in_f <- "SRR616268sub_2.fastq.gz"
out_f <- "hoge5.fastq.gz"
param_trim <- 2
library(Biostrings)
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- width(sread(fastq)) - param_trim
hoge[hoge < 1] <- 1
hoge1 <- DNAStringSet(sread(fastq), start=1, end=hoge)
hoge2 <- BStringSet(quality(quality(fastq)), start=1, end=hoge)
fastq <- ShortReadQ(hoge1, hoge2, id(fastq))
sread(fastq)
quality(fastq)
writeFastq(fastq, out_f, compress=T)
前処理 | トリミング | アダプター配列除去(応用) | QuasR(Gaidatzis_2015)の例題4実行結果ファイルです。
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約71MB; 71,343,605 bytes)です。
paired-endのforward側です。長さはリードによって異なります。
3'側の7 bp分をトリムしています。
現状では、hoge1作成段階で、14023番目のリードのサブセットを抽出できないという理由でこけます。
この塩基配列とクオリティスコア情報は空なので、そこからサブセットを得ようとしているからです。
in_f <- "SRR616268sub_trim2_1.fastq.gz"
out_f <- "hoge6.fastq.gz"
param_trim <- 7
library(Biostrings)
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- width(sread(fastq)) - param_trim
hoge[hoge < 1] <- 1
hoge1 <- DNAStringSet(sread(fastq), start=1, end=hoge)
hoge2 <- BStringSet(quality(quality(fastq)), start=1, end=hoge)
fastq <- ShortReadQ(hoge1, hoge2, id(fastq))
sread(fastq)
quality(fastq)
writeFastq(fastq, out_f, compress=T)
前処理 | トリミング | アダプター配列除去(応用) | QuasR(Gaidatzis_2015)の例題5実行結果ファイルです。
乳酸菌RNA-seqデータSRR616268の最初の100万リード分(約64MB; 63,524,791 bytes)です。
paired-endのreverse側です。長さはリードによって異なります。
3'側の2 bp分をトリムしています。
in_f <- "SRR616268sub_trim2_2.fastq.gz"
out_f <- "hoge7.fastq.gz"
param_trim <- 2
library(Biostrings)
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- width(sread(fastq)) - param_trim
hoge[hoge < 1] <- 1
hoge1 <- DNAStringSet(sread(fastq), start=1, end=hoge)
hoge2 <- BStringSet(quality(quality(fastq)), start=1, end=hoge)
fastq <- ShortReadQ(hoge1, hoge2, id(fastq))
sread(fastq)
quality(fastq)
writeFastq(fastq, out_f, compress=T)
前処理 | フィルタリング | について
ここでは、主に塩基配列データ(リードデータ)の前処理について、特にクオリティの低いリードのフィルタリングを中心にリストアップしています。
発現変動解析を行う前に低発現遺伝子やサンプル間で発現変動のない遺伝子を予め除去するという意味でのフィルタリングについては、
「解析 | 前処理 | について 」のあたりをご覧ください。
前処理 | フィルタリング | PHREDスコアが低い塩基をNに置換
Sanger FASTQ形式ファイルを読み込んで、PHREDスコアが任意の閾値未満の塩基を"N"に置換するやり方を紹介します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge1.fastq"
param1 <- 20
param2 <- "N"
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge1 <- as(quality(fastq), "matrix")
obj <- hoge1 < param1
hoge2 <- DNAString(paste(rep(param2, max(width(fastq))), collapse=""))
hoge3 <- as(Views(hoge2, start=1, end=rowSums(obj)), "DNAStringSet")
fasta <- replaceLetterAt(sread(fastq), obj, hoge3)
fastq <- ShortReadQ(fasta, quality(fastq), id(fastq))
sread(fastq)
writeFastq(fastq, out_f, compress=F)
前処理 | フィルタリング | PHREDスコアが低い配列(リード)を除去
Sanger FASTQ形式ファイルを読み込んで、PHREDスコアが低いリードを除去するやり方を紹介します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
PHREDスコアが20未満のものがリード長に占める割合が0.1以上のリードを除去するやり方です。
(例題のファイル中のリードは全て35bpのリードである。その10%以上ということで実質的にPHREDスコアが閾値未満のものが4塩基以上あるリードはダメということ)
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge1.fastq"
param1 <- 20
param2 <- 0.1
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- as(quality(fastq), "matrix")
obj <- (rowSums(hoge < param1) <= width(fastq)*param2)
fastq <- fastq[obj]
sread(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
PHREDスコアが20以上の塩基数が0.9より多いリードのみ抽出するやり方です。
1と同じことを別の言葉で表現しているだけです。
in_f <- "SRR037439.fastq"
out_f <- "hoge2.fastq"
param1 <- 20
param2 <- 0.9
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- as(quality(fastq), "matrix")
obj <- (rowSums(hoge >= param1) > width(fastq)*param2)
fastq <- fastq[obj]
sread(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
PHREDスコアが20以上の塩基数と配列長に占める割合(%)を出力させるやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge3.txt"
param1 <- 20
library(ShortRead)
fastq <- readFastq(in_f)
hoge <- as(quality(fastq), "matrix")
obj_num <- rowSums(hoge >= param1)
obj_percent <- 100*obj_num/width(fastq)
hoge <- strsplit(as.character(id(fastq)), " ", fixed=TRUE)
description <- sapply(hoge,"[[", 1)
tmp <- cbind(description, width(fastq), obj_num, obj_percent)
colnames(tmp) <- c("description", "read_length", "nucleotide_number", "percentage")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
PHREDスコアが20未満のものがリード長に占める割合が0.8以上のリードを除去するやり方です。
あるチュートリアル(Short Reads Quality Control and Preprocessingの6ページ目)
に書いてあったフィルタリング条件(less than 20 over the 80%)です。
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "SRR037439.fastq"
out_f <- "hoge4.fastq"
param1 <- 20
param2 <- 0.8
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
hoge <- as(quality(fastq), "matrix")
obj <- (rowSums(hoge < param1) <= width(fastq)*param2)
fastq <- fastq[obj]
sread(fastq)
writeFastq(fastq, out_f, compress=F)
前処理 | フィルタリング | ACGTのみからなる配列を抽出
FASTQファイルやFASTAファイルを読み込んで"N"などの文字を含まず、ACGTのみからなる配列のみ抽出して、(multi-)FASTA形式ファイルに出力するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
in_f <- "SRR037439.fastq"
out_f <- "hoge1.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
出力ファイルのdescription行を「kkk_...」に変更するやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge2.fasta"
param1 <- "kkk"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
names(fasta) <- paste(param1, 1:length(fasta), sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
配列中にNを含まないので、入力ファイルと同じ結果が返されることがわかります。
in_f <- "hoge4.fa"
out_f <- "hoge3.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
配列中に"-"を含むcontig_1と配列中に"N"を含むcontig_8, ..., 12が消えていることがわかります。
in_f <- "sample2.fasta"
out_f <- "hoge4.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4.と同じ結果になりますが、やり方を変えています。
in_f <- "sample2.fasta"
out_f <- "hoge5.fasta"
param <- 0
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
6. (multi-)FASTA形式ファイル(sample4.fasta)の場合:
gene_4が消えていることが分かります。
in_f <- "sample4.fasta"
out_f <- "hoge6.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) == hoge)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
sample2.fastaファイル中のcontig_1のように、なぜか文字列以外のcharacter(例:"-")が含まれることがたまにあります。
ここでは、"-"を"N"に変換するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample2.fasta"
out_f <- "hoge1.fasta"
param <- "-"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- chartr(param, "N", fasta)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
前処理 | フィルタリング | ACGT以外の文字数が閾値以下の配列を抽出
(multi-)FASTAファイルを読み込んでACGT以外の文字(実質的にNに相当)数がparam1で指定した閾値以下の配列のみ抽出するやり方を示します。この値を0にすれば、ACGTのみからなる配列の抽出を行うのと同じ意味になります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. FASTA形式ファイル(sample2.fasta)を読み込んでACGTのみからなる配列を抽出したい(Nが0個に相当)場合:
in_f <- "sample2.fasta"
out_f <- "hoge1.fasta"
param1 <- 0
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param1
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. FASTA形式ファイル(sample2.fasta)を読み込んでNが1個含むものまで許容してフィルタリングしたい場合:
in_f <- "sample2.fasta"
out_f <- "hoge2.fasta"
param1 <- 1
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param1
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. FASTA形式ファイル(sample2.fasta)を読み込んでNが1個含むものまで許容してフィルタリングしたい場合:
QuasRパッケージ中のpreprocessReads関数を用いるやり方です。
in_f <- "sample2.fasta"
out_f <- "hoge3.txt"
param1 <- 1
library(QuasR)
out <- preprocessReads(filename=in_f,
outputFilename=out_f,
minLength=0,
nBases=param1)
out
前処理 | フィルタリング | 重複のない配列セットを作成
ユニークな配列からなるセットにするやり方を紹介します。ちなみに、ユニークとはいっても例えば"AACGTTGCA"と"AACGTTGCAG"は独立な配列として取り扱われます。
出力ファイルのdescription行は「kkk_X_Y」に変更しています。重複配列同士のdescription行が異なっていた場合に、どれを使うかという問題があることと、description行部分の記述をすっきりさせて、Xに相当する部分に配列のシリアル番号、そしてYに相当する部分にその配列の出現回数を示すようにしています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
contig_8, 11, 12の三つの配列がまとめられていることなどがわかります。
in_f <- "sample2.fasta"
out_f <- "hoge1.fasta"
param1 <- "kkk"
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- tables(fasta, n=length(unique(fasta)))
fasta <- DNAStringSet(names(hoge$top))
fasta
names(fasta) <- paste(param1, 1:length(fasta), hoge$top, sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
in_f <- "SRR037439.fastq"
out_f <- "hoge2.fasta"
param1 <- "kkk"
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fastq")
fasta
hoge <- tables(fasta, n=length(unique(fasta)))
fasta <- DNAStringSet(names(hoge$top))
fasta
names(fasta) <- paste(param1, 1:length(fasta), hoge$top, sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
配列中にNを含まず重複配列も存在しないので、順番のみ入れ替わった結果が返されていることがわかります。
in_f <- "hoge4.fa"
out_f <- "hoge3.fasta"
param1 <- "kkk"
library(ShortRead)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- tables(fasta, n=length(unique(fasta)))
fasta <- DNAStringSet(names(hoge$top))
fasta
names(fasta) <- paste(param1, 1:length(fasta), hoge$top, sep="_")
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
前処理 | フィルタリング | 指定した長さ以上の配列を抽出
FASTA形式やFASTQ形式ファイルを入力として、指定した配列長以上の配列を抽出するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. multi-FASTAファイル(hoge4.fa)の場合:
イントロ | 一般 | ランダムな塩基配列を作成の4.を実行して得られたファイルです。
in_f <- "hoge4.fa"
out_f <- "hoge1.fasta"
param_length <- 50
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(width(fasta) >= param_length)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
in_f <- "sample2.fasta"
out_f <- "hoge2.fasta"
param_length <- 16
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
table(width(fasta))
obj <- as.logical(width(fasta) >= param_length)
fasta <- fasta[obj]
fasta
table(width(fasta))
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. FASTQ形式ファイル(sample2.fastq)を読み込んでFASTQ形式で保存する場合:
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
in_f <- "sample2.fastq"
out_f <- "hoge3.fastq"
param_length <- 26
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
table(width(fastq))
obj <- as.logical(width(fastq) >= param_length)
fastq <- fastq[obj]
sread(fastq)
table(width(fastq))
writeFastq(fastq, out_f, compress=F)
4. FASTQ形式ファイル(sample2.fastq)を読み込んでFASTA形式で保存する場合:
in_f <- "sample2.fastq"
out_f <- "hoge4.fasta"
param_length <- 26
library(ShortRead)
fastq <- readFastq(in_f)
fasta <- sread(fastq)
fasta
table(width(fasta))
obj <- as.logical(width(fasta) >= param_length)
fasta <- fasta[obj]
fasta
table(width(fasta))
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
前処理 | フィルタリング | 任意のリード(サブセット)を抽出
FASTA形式やFASTQ形式ファイルを入力として、任意の配列(リード)を抽出するやり方を示します。
ここのコードをテンプレートにして、マッピングなどを行う際に動作確認用として指定したリード数からなるサブセットを作成できます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. multi-FASTAファイル(hoge4.fa)の場合:
イントロ | 一般 | ランダムな塩基配列を作成の4.を実行して得られたものです。
最初の3リードを抽出するやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge1.fasta"
param <- 3
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- 1:param
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
2. multi-FASTAファイル(hoge4.fa)の場合:
(全部で4リードからなることが既知という前提で)2-4番目のリードを抽出するやり方です。
in_f <- "hoge4.fa"
out_f <- "hoge2.fasta"
param_range <- 2:4
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- param_range
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
3. multi-FASTAファイル(hoge4.fa)の場合:
1.と同じで最初の3リードを抽出するやり方ですが、表現方法が異なります。
in_f <- "hoge4.fa"
out_f <- "hoge3.fasta"
param_range <- 1:3
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- param_range
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
4. FASTQ形式ファイル(sample2.fastq)を読み込んでFASTQ形式で保存する場合:
writeFastq関数のデフォルトオプションはcompress=Tで、gzip圧縮ファイルを出力します。
ここではcompress=Fとして非圧縮ファイルを出力しています。
(全部で187リードからなることが既知という前提で)5-20番目のリードを抽出するやり方です。
in_f <- "sample2.fastq"
out_f <- "hoge4.fastq"
param_range <- 5:20
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
obj <- param_range
fastq <- fastq[obj]
sread(fastq)
writeFastq(fastq, out_f, compress=F)
5. FASTQ形式ファイル(sample2.fastq)を読み込んでFASTQ形式で保存する場合:
(全部で187リードからなることが既知という前提でそれよりも少ない)30リード分をランダムに非復元抽出するやり方です。
writeFastq関数実行時にcompress=Fとしてgzip圧縮前のファイルを出力しています
in_f <- "sample2.fastq"
out_f <- "hoge5.fastq"
param <- 30
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
set.seed(1010)
obj <- sample(1:length(fastq), param, replace=F)
fastq <- fastq[sort(obj)]
sread(fastq)
writeFastq(fastq, out_f, compress=F)
6. FASTQ形式ファイル(sample2.fastq)を読み込んでFASTQ形式で保存する場合:
(全部で187リードからなることが既知という前提でそれよりも少ない)30リード分をランダムに非復元抽出するやり方です。
5.と実質的に同じですが、gzip圧縮ファイルとして保存するやり方です。Macintoshではうまくいかないかもしれません。
in_f <- "sample2.fastq"
out_f <- "hoge6.fastq.gz"
param <- 30
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
set.seed(1010)
obj <- sample(1:length(fastq), param, replace=F)
fastq <- fastq[sort(obj)]
sread(fastq)
writeFastq(fastq, out_f, compress=T)
7. FASTQ形式ファイル(sample2.fastq)を読み込んでFASTA形式で保存する場合:
(全部で187リードからなることが既知という前提でそれよりも少ない)30リード分をランダムに非復元抽出するやり方です。
in_f <- "sample2.fastq"
out_f <- "hoge7.fasta"
param <- 30
library(ShortRead)
fastq <- readFastq(in_f)
fasta <- sread(fastq)
names(fasta) <- id(fastq)
fasta
set.seed(1010)
obj <- sample(1:length(fasta), param, replace=F)
fasta <- fasta[sort(obj)]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
8. small RNA-seqのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)を読み込んでFASTQ形式で保存する場合:
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られた
カイコsmall RNA-seqデータ(Nie et al., BMC Genomics, 2013)です。
入力ファイルサイズは400Mb弱、11,928,428リードです。
この中から100000リード分をランダムに非復元抽出した結果をgzip圧縮なしで出力しています。
出力ファイルはSRR609266_sub.fastqと同じもの(100000リード; 約16MB)になります。
Macintoshではうまくいかないかもしれません。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge8.fastq"
param <- 100000
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
set.seed(1010)
obj <- sample(1:length(fastq), param, replace=F)
fastq <- fastq[sort(obj)]
id(fastq)
writeFastq(fastq, out_f, compress=F)
9. small RNA-seqのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)を読み込んでFASTQ形式で保存する場合:
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られた
カイコsmall RNA-seqデータ(Nie et al., BMC Genomics, 2013)です。
入力ファイルサイズは400Mb弱、11,928,428リードです。
この中から最初の100000リード分をgzip圧縮なしで出力しています。
Macintoshではうまくいかないかもしれません。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge9.fastq"
param <- 100000
library(ShortRead)
fastq <- readFastq(in_f)
id(fastq)
fastq <- fastq[1:param]
id(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
(全部で500リードからなることが既知という前提で)5-20番目のリードを抽出するやり方です。
in_f <- "SRR037439.fastq"
out_f <- "hoge10.fastq"
param_range <- 5:20
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
obj <- param_range
fastq <- fastq[obj]
sread(fastq)
writeFastq(fastq, out_f, compress=F)
SRR037439から得られるFASTQファイルの最初の2,000行分を抽出したMAQC2 brainデータです(Bullard et al., 2010)。
(全部で500リードからなることが既知という前提で)30リード分をランダムに非復元抽出するやり方です。
writeFastq関数実行時にcompress=Fとしてgzip圧縮前のファイルを出力しています
in_f <- "SRR037439.fastq"
out_f <- "hoge11.fastq"
param <- 30
library(ShortRead)
fastq <- readFastq(in_f)
sread(fastq)
set.seed(1010)
obj <- sample(1:length(fastq), param, replace=F)
fastq <- fastq[sort(obj)]
sread(fastq)
writeFastq(fastq, out_f, compress=F)
前処理 | フィルタリング | 指定した長さの範囲の配列
指定した長さの範囲の配列を抽出するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
長さの範囲が20-30bpの配列を抽出するやり方を示します。
in_f <- "sample2.fasta"
out_f <- "hoge1.fasta"
param_range <- 20:30
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- (width(fasta) >= min(param_range)) & (width(fasta) <= max(param_range))
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
前処理 | フィルタリング | 任意のIDを含む配列を抽出
二つのファイル(multi-FASTA形式ファイルとIDリストファイル)を読み込んで、IDリストファイル中のRefSeq IDに対応する配列のサブセットを抽出するやり方を示します。
multi-FASTAファイルのdescription行の記述中にIDリストファイル中の文字列が存在することが大前提であることはいうまでもありませんが一応言っときます。
また、同一文字列としてうまく認識させるために、(description行の記述の中からIDリスト中の文字列と完全一致させるべく)
description部分の記述をうまく分割(例:"_up_"など)してIDと一致する部分のみにする必要があり、このあたりは、自分でdescription行を眺めて適宜改変しなければなりません。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
(multi-FASTAファイルのほうのdescription行を"_up_"を区切り文字として分割し、分割後の1番目の要素を抽出することでIDリストとの対応付けを可能としている)
in_f1 <- "rat_upstream_1000.fa"
in_f2 <- "sample_IDlist1.txt"
out_f <- "hoge1.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
hoge <- strsplit(names(fasta), "_up_", fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", 1))
names(fasta) <- hoge2
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
(multi-FASTAファイルのほうのdescription行を"|"を区切り文字として分割し、分割後の4番目の要素を抽出することでIDリストとの対応付けを可能としているつもりだが、バージョン番号が残っているのでうまくいかない例)
in_f1 <- "sample_100.fasta"
in_f2 <- "sample_IDlist2.txt"
out_f <- "hoge2.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
hoge <- strsplit(names(fasta), "|", fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", 4))
names(fasta) <- hoge2
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
(バージョン番号つきRefSeq IDをさらに"."を区切り文字として分割し、分割後の1番目の要素を抽出することでIDリストとの対応付けを可能としている)
in_f1 <- "sample_100.fasta"
in_f2 <- "sample_IDlist2.txt"
out_f <- "hoge3.fasta"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
keywords <- readLines(in_f2)
fasta
hoge <- strsplit(names(fasta), "|", fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", 4))
hoge3 <- strsplit(hoge2, ".", fixed=TRUE)
hoge4 <- unlist(lapply(hoge3, "[[", 1))
names(fasta) <- hoge4
fasta
obj <- is.element(names(fasta), keywords)
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
前処理 | フィルタリング | Illuminaのpass filtering
ここはまだ未完成です。。。
二つのpaired-endファイル(read1.fqとread2.fq)があるという前提で二つのファイル中で両方ともpass filteringフラグが1となっているものを抽出するやり方を示します。ここで読み込む二つのファイルは同一行に同一IDが位置するファイルであるという前提で行います。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f1 <- "read1.fq"
in_f2 <- "read2.fq"
out_f1 <- "read1_pf.fastq"
out_f2 <- "read2_pf.fastq"
library(ShortRead)
reads1 <- readFastq(in_f1)
hoge <- strsplit(as.character(id(reads1)), "/", fixed=TRUE)
hoge2 <- sapply(hoge,"[[", 1)
hoge3 <- strsplit(hoge2, ":", fixed=TRUE)
pass_filter1 <- sapply(hoge3,"[[", length(hoge3[[1]]))
tmp_serial1 <- hoge2
reads2 <- readFastq(in_f2)
hoge <- strsplit(as.character(id(reads2)), "/", fixed=TRUE)
hoge2 <- sapply(hoge,"[[", 1)
hoge3 <- strsplit(hoge2, ":", fixed=TRUE)
pass_filter2 <- sapply(hoge3,"[[", length(hoge3[[1]]))
tmp_serial2 <- hoge2
flag <- 0
if(length(tmp_serial1) == length(tmp_serial2)){
print("Step1:OK")
flag <- 1
if(length(tmp_serial1) == sum(tmp_serial1 == tmp_serial2)){
print("Step2:OK")
flag <- 2
}else{
print("Step2:IDs between two paired-end files are not ideltical!")
}
}else{
print("Step1:The two files are not paired-end!")
}
if(flag == 2){
hoge_flag <- as.integer(pass_filter1) + as.integer(pass_filter2)
length(hoge_flag)
sum(hoge_flag == 2)
writeFastq(reads1[hoge_flag == 2], out_f1)
writeFastq(reads2[hoge_flag == 2], out_f2)
}
前処理 | フィルタリング | GFF/GTF形式ファイル
QuasRパッケージを用いてゲノムへのマッピング結果からカウント情報を得たいときに、 "TxDb"という形式のオブジェクトを利用する必要があります。
この形式のオブジェクトは、Rで提供されていない生物種の遺伝子アノテーションパッケージ以外のものについても、GFF/GTF形式のファイルがあれば、
それを入力としてTxDbオブジェクトを作成するための関数(GenomicFeaturesパッケージ中のmakeTxDbFromGFF関数)が提供されています。
しかし現実には、GFF/GTF形式ファイルは提供元によって微妙に異なるため、エラーを吐いてmakeTxDbFromGFF関数を実行できない場合が多いです。
それゆえイントロ | 一般 | 任意の文字列を行の最初に挿入のような小細工をして、
染色体名を同じ文字列として認識できるようにしたファイル(human_annotation_sub2.gtf)を用意する必要があるのですが、それでもまだだめな場合があります。
それは、EnsemblのFTPサイトから提供されているGFF/GTFファイル中には、
例えばヒトの場合「chr1, chr2, ...chr22, chrX, chrY, chrMT」以外に"HSCHR6_MHC_APD"や"HSCHR6_MHC_COX"などの記述があるためです。
例えばリファレンス配列を"BSgenome.Hsapiens.UCSC.hg19"に指定して行った場合には、"HSCHR6_MHC_APD"がリファレンス配列中に存在しないため、
GTFファイルに存在する"HSCHR6_MHC_APD"コンティグ?!上の特定の領域にマップされたリード数をカウントできるはずもなく、結果的に「そんなものはない!」とmakeTxDbFromGFF関数実行中に怒られるわけです。
この問題への最も合理的な対処法は、GFF/GTFファイルの中から、自分がマッピングに用いたリファレンス配列中の染色体名と同じ文字列の行だけを抽出した新たなGFF/GTFファイルを作成することです。
したがって、ここではGFF/GTFファイルとリファレンス配列を読み込んで、リファレンス配列中に存在する染色体名を含む行だけを抽出するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. ヒトゲノム配列("BSgenome.Hsapiens.UCSC.hg19")中の染色体名と一致する遺伝子アノテーション情報のみGTFファイル
(human_annotation_sub2.gtf)から抽出したい場合:
in_f1 <- "human_annotation_sub2.gtf"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f <- "hoge1.txt"
data <- read.table(in_f1, header=FALSE, sep="\t", quote="")
dim(data)
param <- in_f2
library(param, character.only=T)
tmp <- ls(paste("package", param, sep=":"))
hoge <- eval(parse(text=tmp))
keywords <- seqnames(hoge)
keywords
obj <- is.element(as.character(data[,1]), keywords)
out <- data[obj,]
dim(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
前処理 | フィルタリング | 組合せ | ACGTのみ & 指定した長さの範囲の配列
フィルタリングを組み合わせるやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
ACGTのみからなる配列を抽出したのち、さらに指定した長さの範囲(20-30bp)の配列を抽出するやり方を示します。
in_f <- "sample2.fasta"
out_f <- "hoge1.fasta"
param_nBases <- 0
param_range <- 20:30
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
#hoge <- rowSums(alphabetFrequency(DNAStringSet(fasta))[,1:4])
hoge <- apply(as.matrix(alphabetFrequency(DNAStringSet(fasta))[,1:4]), 1, sum)
obj <- (width(fasta) - hoge) <= param_nBases
fasta <- fasta[obj]
fasta
obj <- (width(fasta) >= min(param_range)) & (width(fasta) <= max(param_range))
fasta <- fasta[obj]
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
前処理 | フィルタリング | paired-end | 配列長とN数 | QuasR(Gaidatzis_2015)
QuasRパッケージを用いて、
リード数が同じpaired-endデータの2つのファイルを入力として、配列長やN数でのフィルタリング例を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
前処理 | トリミング | アダプター配列除去(応用) | QuasR(Gaidatzis_2015)の例題4と5の実行結果ファイルで、paired-endデータです。
SRR616268sub_trim2_1.fastq.gzは、1,000,000リード、71,343,605 bytes (約71MB)です。
SRR616268sub_trim2_2.fastq.gzは、1,000,000リード、63,524,791 bytes (約64MB)です。
tooShortが107、tooManyNが1,551リードあったことがわかります。結果として、998,431リードからなるファイルが出力されています。
尚、998,431 + 107 + 1,551 = 1,000,089リードであったことから、
tooShort且つtooManyNのものの和集合(union)は(1,000,000 - 998,431) = 1,569リードなのだろうと解釈しました。
約2分。
in_f1 <- "SRR616268sub_trim2_1.fastq.gz"
in_f2 <- "SRR616268sub_trim2_2.fastq.gz"
out_f1 <- "hoge1_1.fastq.gz"
out_f2 <- "hoge1_2.fastq.gz"
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f1,
filenameMate=in_f2,
outputFilename=out_f1,
outputFilenameMate=out_f2,
nrec=param_nrec)
res
許容するN数(param_nBases)と最短配列長(param_minLength)を明示的に与えるやり方です。
tooShortが119、tooManyNが1,762リードあったことがわかります。結果として、998,208リードからなるファイルが出力されています。
例題6に比べて若干生き残るリード数が減るのは、param_nBases(デフォルトは2)と
param_minLength(デフォルトは14)での条件がデフォルトよりも若干厳しめ
(1と18)だからです。約2分。
in_f1 <- "SRR616268sub_trim2_1.fastq.gz"
in_f2 <- "SRR616268sub_trim2_2.fastq.gz"
out_f1 <- "hoge2_1.fastq.gz"
out_f2 <- "hoge2_2.fastq.gz"
param_nBases <- 1
param_minLength <- 18
param_nrec <- 500000
library(QuasR)
res <- preprocessReads(filename=in_f1,
filenameMate=in_f2,
outputFilename=out_f1,
outputFilenameMate=out_f2,
nBases=param_nBases,
minLength=param_minLength,
nrec=param_nrec)
res
前処理 | フィルタリング | paired-end | 共通リード抽出 | ShortRead(Morgan_2009)
ShortReadパッケージを用いて、
ファイル内での配列長とリード数が異なるpaired-endデータの2つのファイルを入力として、
両方に共通して存在するリードのみ抽出するやり方を示します。
QuasRは入力ファイルのリード数が揃ってないと受けつけないので、
ShortReadパッケージで別々に読み込む戦略を採用しています。
前処理 | トリミング | アダプター配列除去(基礎) | QuasR(Gaidatzis_2015)
の例題7と8の実行結果ファイルで、100万リード弱のpaired-endデータです。
SRR616268sub_trim_1.fastq.gzは、998,658リード、71,227,695 bytes (約71MB)です。
SRR616268sub_trim_2.fastq.gzは、999,136リード、63,442,904 bytes (約63MB)です。
前処理のところで長ったらしい作業をしているのは、FASTQファイル中のdescription行でリードのIDの対応付けがそのままではできないので、
対応付け可能な部分文字列を抽出しているためです。
イントロ | NGS | 読み込み | FASTQ形式 | description行の記述を整形のコードをテンプレートにしています。
共通リードは998,428個です。リード数がほとんど減っていないにもかかわらず、ファイルサイズが(71,227,695 bytes -> 63,446,240 bytes;
63,442,904 bytes -> 56,019,410 bytes)と大幅に減っているのは、description部分の記述内容が減っていることに起因します。
in_f1 <- "SRR616268sub_trim_1.fastq.gz"
in_f2 <- "SRR616268sub_trim_2.fastq.gz"
out_f1 <- "hoge1_1.fastq.gz"
out_f2 <- "hoge1_2.fastq.gz"
library(ShortRead)
fastq1 <- readFastq(in_f1)
fastq2 <- readFastq(in_f2)
head(id(fastq1), n=4)
head(id(fastq2), n=4)
fastq <- fastq1
hoge <- strsplit(as.character(id(fastq)), " ", fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", 1))
fastq <- ShortReadQ(sread(fastq), quality(fastq), description)
head(id(fastq), n=4)
fastq1 <- fastq
fastq <- fastq2
hoge <- strsplit(as.character(id(fastq)), " ", fixed=TRUE)
description <- BStringSet(sapply(hoge,"[[", 1))
fastq <- ShortReadQ(sread(fastq), quality(fastq), description)
head(id(fastq), n=4)
fastq2 <- fastq
common <- intersect(id(fastq1), id(fastq2))
length(fastq1)
length(fastq2)
length(common)
obj1 <- is.element(as.character(id(fastq1)), as.character(common))
obj2 <- is.element(as.character(id(fastq2)), as.character(common))
writeFastq(fastq1[obj1], out_f1, compress=T)
writeFastq(fastq2[obj2], out_f2, compress=T)
file.size(in_f1)
file.size(in_f2)
file.size(out_f1)
file.size(out_f2)
アセンブル | について
アセンブルという言葉がなかなか理解しずらい人は、単純に「多重配列アラインメント(multiple sequence alignment)を行って一本一本の配列だけからでは到底到達できないような長い配列(これをコンセンサス配列という)を作成すること」だと解釈することで差し支えないと思います。
出力ファイル形式はFASTAがデファクトスタンダードです。
アセンブルによって、まず一本一本のリードをコンティグ(contigs)にまとめます。
そしてコンティグの並び(order or orientation)やコンティグ間のギャップサイズ("N"の数で表現)をスカッフォールド(スキャッフォールド;scaffolds)で表します。
(別の言葉でいうと、scaffoldsは複数のコンティグの並びがどうなっているかを表したもの or まとめたもの、です。)
一般にFASTA形式のアセンブルされた配列のファイルとともに、配列に関するいくつかの統計値も得られます。
最大配列長(maximum length)、平均配列長(average length)、全配列を結合して得られる長さ(combined total length)、N50などです。
ちなみにこのN50というのは、「最も長い配列から順番に連結していったときにcombined total lengthの50%になったときの配列の長さ」です。
「combined total lengthが50%となるような最小数の配列集合のうち最も小さい配列の長さ」という言い方もできます。
length-weighted medianという表現もなされるようです。
アセンブリを行う際に問題となるのは「リピート配列の取り扱い」や「アセンブルの際に設定する閾値」です。
前者については、ペアードエンド(paired-end)の一部がリピート配列の末端部分だったり、ペアードエンドの片方がリピート配列そのものであればリピート配列の素性がかなりわかります。
後者については、いわゆる感度・特異度の議論と本質的に同じで、combined total lengthをより長くする(つまり感度を上げたい)ためには多少ミスアセンブル(特異度が下がること; chimeric assembly)を許容する必要があります。
デノボゲノムアセンブリ(de novo genome assembly):
ゲノムアセンブリの手順は、以下に示すように大きく4つのステージに分けられるそうです(El-Metwally et al., PLoS Comput Biol., 2013):
- 前処理(pre-processing filtering; error correction)
- グラフ構築(graph construction process)
- グラフ簡易化(graph simplification process)
- 後処理(post-processing filtering)
1. 前処理(pre-processing filtering; error correction):
ここで行う処理は、塩基置換(substitution; mismatch)、インデル(indels; insertion/deletion)、曖昧な塩基(N)を含むリードの除去や補正です。
基本的な戦略は「エラーの頻度は低い」ですが、原理的に高頻度で出現するリピート配列の悪影響を受けるようです。
シークエンスエラー(sequencing error)と多型(polymorphism)の違いはグラフ構築後でないとわからないので、2.のグラフ構築ステップ時に行う場合もあるようです。
また、アセンブルプログラムの中に完全に組み込まれていたりなど切り分けは若干ややこしいですがざっとリストアップしておきます。
エラー補正プログラムとほぼ同義です。ちなみに、初期のアセンブラはこのエラー補正ステップがないようです(El-Metwally et al., PLoS Comput Biol., 2013)。
この理由は、ABI3730のような800bp程度まで読めるロングリードのアセンブリの場合は配列一致部分も長い(long overlap)ので、一致部分に多少のエラーを含もうが影響は限定的だったからです。
エラー補正は大きく4つのアプローチに分けられるそうです:K-spectrum (k-mer), Suffix Tree/Array (STA), Multiple Sequence Alignment (MSA), Hybrid。
- FreClu:Qu et al., Genome Res., 2009
- SHREC(STA correction):Schröder et al., Bioinformatics, 2009
- Hybrid SHREC(Hybrid correction):Salmela L., Bioinformatics, 2010
- Reptile(k-mer correction):Yang et al., Bioinformatics, 2010
- EDAR:Zhao et al., J Comput Biol., 2010
- Quake(k-mer correction):Kelley et al., Genome Biol., 2010
- HiTEC(STA correction):Ilie et al., Bioinformatics, 2011
- REDEEM:Yang et al., BMC Bioinformatics, 2011
- DecGPU:Liu et al., BMC Bioinformatics, 2011
- Coral(MSA correction):Salmela et al., Bioinformatics, 2011
- ECHO(MSA correction):Kao et al., Genome Res., 2011
- Hammer(k-mer correction):Medvedev et al., Bioinformatics, 2011
- PBcR(Hybrid correction):Koren et al., Nat Biotechnol., 2012
- KEC and ET:Skums et al., BMC Bioinformatics, 2012
- BayesHammer:Nikolenko et al., BMC Genomics, 2013
- SEECER:Le et al., Nucleic Acids Res., 2013
- RACER:Ilie et al., Bioinformatics, 2013
- CorQ:Iyer et al., PLoS One, 2013
- Sleep's method:Sleep et al., BMC Bioinformatics, 2013
- BLESS:Heo et al., Bioinformatics, 2014
- HECTOR:Wirawan et al., BMC Bioinformatics, 2014
- Blue:Greenfield et al., Bioinformatics, 2014
2. グラフ構築(graph construction process):
ここで行うのは、前処理後のリードを用いてリード間のオーバーラップ(overlap)を頼りにつなげていく作業です。
シークエンスエラー(sequencing error)と多型(polymorphism)の違いを見るべく、グラフ構築時にエラー補正を行うものもあります。
おそらく全てのアセンブルプログラムはグラフ理論(Graph theory)を用いています。
一筆書きの問題をグラフ化して解くような学問です。
それらはさらに4つのアプローチに大別できます: Overlap (OLC), de Bruign (k-mer), Greedy, Hybrid。
2-1. Overlap-Layout-Consensus (OLC)アプローチ。アセンブル問題をハミルトンパス(Hamiltonian path)問題として解く、ABI3730 sequencerの頃から存在する伝統的な方法。
overlap, layout, and consensusの3つのステップからなるためOLCと略される。
454など比較的長い配列(数百塩基程度)のアセンブルを目的としたものが多いようですが、最近ではSGAやReadjoinerなどショートリードにもうまく対応したものが出てきているようです。
- Celera Assembler(OLC graph):Myers et al., Science, 2000
- ARACHNE(OLC graph):Batzoglou et al., Genome Res, 2002
- PCAP(OLC graph):Huang et al., Genome Res., 2003
- Newbler(OLC graph):Margulies et al., Nature, 2005
- Edena(OLC graph):Hernandez et al., Genome Res., 2008
- CABOG(MSA correction; OLC graph):Miller et al., Bioinformatics, 2008
- SHORTY(OLC graph; scaffolder):Hossain et al., BMC Bioinformatics, 2009
- Forge(OLC graph):Diguistini et al., Genome Biol., 2009
- SGA(k-mer correction; OLC graph; scaffolder):Simpson et al., Genome Res., 2012
- Readjoiner(k-mer correction; OLC graph):Gonnella et al., BMC Bioinformatics, 2012
- Fermi(OLC graph):Li H., Bioinformatics, 2012
2-2. de Bruign (k-mer)アプローチ。これは、de Bruijn graph (DBG)中のオイラーパス探索(DBG approach; Eulerian approach; Euler approach; Eulerian path approach)に基づくもので、
グラフは頂点(ノード; nodes or vertices; 一つ一つの配列に相当)と辺(エッジ; edges or arcs)で表されますが、
リードを1塩基ずつずらして全ての可能なk-mer (all possible fixed k length strings; k個の連続塩基のことでkは任意の正の整数)を生成し各k-merをノードとした有向グラフ(k-merグラフ)を作成します。
全リードに対して同様の作業を行い、完全一致ノードをマージして得られるグラフがDBGです。
そしてこのグラフは各エッジを一度だけ通るオイラーパス(Eulerian path)をもつことが分かっているので、あとは既知のオイラーパス問題専用アルゴリズムを適用するというアプローチです。
どのk-merの値を用いればいいかはなかなか難しいようですが、KmerGenie
(Chikh et al., Bioinformatics, 2014)という最適なkの値を見積もってくれるプログラムもあるようです。
- Velvet(k-mer graph; scaffolder):Zerbino and Birney, Genome Res., 2008
- EULER-SR(k-mer correction; k-mer graph; scaffolder):Chaisson et al., Genome Res., 2009
- ABySS(k-mer graph):Simpson et al., Genome Res., 2009
- ALLPATHS 2(k-mer correction; k-mer graph; scaffolder):Maccallum et al., Genome Biol., 2009
- SOAPdenovo(k-mer correction; k-mer graph; scaffolder):Li et al., Genome Res., 2010
- ALLPATHS-LG(k-mer correction; k-mer graph; scaffolder):Gnerre et al., Proc Natl Acad Sci U S A, 2011
- SparseAssembler(k-mer graph):Ye et al., BMC Bioinformatics, 2012
- Platanus(k-mer correction; k-mer graph; scaffolder):Kajitani et al., Genome Res., 2014
2-3. Greedyアプローチ。貪欲法(Greedy algorithm)に基づくものですが、paired-endなどのリード全体の関係を考慮しないため、
あまり広くは使われていないようです。single-endリードを入力とし、比較的小さいサイズのゲノムに対して適しているとのことです
(El-Metwally et al., PLoS Comput Biol., 2013)。
2-4. Hybridアプローチ。上記の異なる方法を組み合わせたhybridと、異なるシーケンサーを組み合わせたhybridの2種類あります。おそらく後者がhybrid assemblyと言われるものに相当します。
- (454 and Illumina):Reinhardt et al., Genome Res., 2009
- (454 and Illumina):Aury et al., BMC Genomics, 2008
- Taipan(OLC and greedy):Schmidt et al., Bioinformatics, 2009
- (OLC and k-mer):Cerdeira et al., J Microbiol Methods, 2011
- (454, SOLiD and Illumina):Wang et al., BMC Syst Biol., 2012
3. グラフ簡易化(graph simplification process):
ここで行うのは、グラフ構築後に複雑化したグラフをシンプルにしていく作業です。連続したノード(consecutive node) やバブル (bubble)のマージ作業に相当します。
Dead endの除去やリピート領域で形成されるX-cutを2つのパスに切り分ける作業なども含みます。
4. 後処理(post-processing filtering):
ここで行うのは、簡易化後のグラフを一筆書き(渡り歩く、とか横切る、というイメージでよい;これがトラバース)してコンティグを得る作業です。
paired-endリード情報を用いて、コンティグ同士を連結させたスーパーコンティグ(super-contigs)またはスカッフォールド(scaffolds)構築や、ミスアセンブリの同定も含みます。
2.のアセンブラの中にscaffolderを書いてあるのはscaffolding moduleを持っているものたちです(El-Metwally et al., PLoS Comput Biol., 2013)。
記載もれもあるとは思います。以下に示すのは、scaffoldingのみを単独で行うプログラムたちです。
- Bambus:Pop et al., Genome Res., 2004
- SOPRA:Dayarian et al., BMC Bioinformatics, 2010
- SSPACE:Boetzer et al., Bioinformatics, 2011
- Opera:Gao et al., J Comput Biol., 2011
- MIP Scaffolder:Salmela et al., Bioinformatics, 2011
- GRASS:Gritsenko et al., Bioinformatics, 2012
- SCARPA:Donmez et al., Bioinformatics, 2013
- L_RNA_scaffolder:Xue et al., BMC Genomics, 2013
- 手法比較論文:Hunt et al., Genome Biol., 2014
評価体系:
アセンブリの精度評価(確からしさの見積もり)は一般に難しいようですが、メイトペア(mate pair)による制約(constraint)を一般に利用してアセンブルしますので、
それを満たしている(satisfaction)のがどの程度あるかやその制約に反した(vaiolation)結果がどの程度あったかということを精度評価に用いるというやり方も提案されています。
リファレンスとなるゲノム配列が既知の場合にはそれとの比較が有用だろうと思いますが、このあたりは私の守備範囲ではありません。
どのアセンブラがいいかについての評価を行う枠組みもあるようです。
トランスクリプトーム用アセンブラ:
どの方法も基本的にアセンブルする(配列同士を連結する)かどうかの判断基準として、k-mer (k個の連続塩基が)一致しているかどうかで判断しますが、
ゲノム用を改良して作成した初期のもののは、このkについて複数の値を利用するという戦略(de novo transcriptome assembly with multiple k-mer values)をとっているようです。
背景としては、ゲノム配列のアセンブリはゲノム全体でcoverageが一般に一定である(or 一定であることを仮定している)のに対して、
トランスクリプトームは転写物によってかなりcoverageが異なります。これは、遺伝子ごとに発現レベルが違うということをcoverageという別の言葉で説明しているだけです。
一般に、kの値を大きくすると高発現遺伝子(高発現転写物)からなるより長い"contig"が得られ、他方kを小さくすると低発現遺伝子がちゃんと"contig"として得られるといった感じになります。
もう少し別な言い方をすると、「kを大きくするとより長いcontig (transcript fragment; 転写物断片)が得られ、kを小さくすると転写物断片がさらに断片化されたようなものが得られる」というイメージです。
kの範囲についてですが、例えばTrans-ABySSはリードの長さがLだとするとL/2からL-1の範囲にしています。
ちなみに一般にkは奇数(odd number)を採用しますが、これはパリンドローム(palindrome)を回避するためらしいです。
例えば5'-ACGTTGCA-3'という配列でk=8を考えてみるとこの相補鎖も全く同じ(そういう配列がパリンドロームだから)で混乱してしまいますが、
kが例えば7や9のような奇数なら相補鎖にマッチすることはないのでパリンドローム問題にぶち当たることはないわけです。
アセンブル | ゲノム用
Rパッケージはおそらくありません。
プログラム:
- Phusion:Mullikin and Ning, Genome Res., 2003
- PCAP:Huang et al., Genome Res., 2003
- Newbler:Margulies et al., Nature, 2005
- SSAKE:Warren et al., Bioinformatics, 2007
- Minimus:Sommer et al., BMC Bioinformatics, 2007
- VCAKE:Jeck et al., Bioinformatics, 2007
- SHARCGS:Dohm et al., Genome Res., 2007
- Edena:Hernandez et al., Genome Res., 2008
- ALLPATHS:Butler et al., Genome Res., 2008
- Velvet:Zerbino and Birney, Genome Res., 2008
- CABOG:Miller et al., Bioinformatics, 2008
- EULER-SR:Chaisson et al., Genome Res., 2009
- SHORTY:Hossain et al., BMC Bioinformatics, 2009
- QSRA:Bryant et al., BMC Bioinformatics, 2009
- ABySS:Simpson et al., Genome Res., 2009
- Taipan:Schmidt et al., Bioinformatics, 2009
- Forge:Diguistini et al., Genome Biol., 2009
- ALLPATHS 2:Maccallum et al., Genome Biol., 2009
- SOAPdenovo:Li et al., Genome Res., 2010
- ALLPATHS-LG:Gnerre et al., Proc Natl Acad Sci U S A, 2011
- SGA:Simpson et al., Genome Res., 2012
- Mapsembler:Peterlongo and Chikhi, BMC Bioinformatics, 2012
- IDBA-UD:Peng et al., Bioinformatics, 2012
- SPAdes:Bankevich et al., J Comput Biol., 2012
- SparseAssembler:Ye et al., BMC Bioinformatics, 2012
- Readjoiner:Gonnella et al., BMC Bioinformatics, 2012
- Fermi:Li H., Bioinformatics, 2012
- MetaVelvet(メタゲノム用):Namiki et al., Nucleic Acids Res., 2012
- TIGER:Wu et al., BMC Bioinformatics, 2012
- CloudBrush:Chang et al., BMC Genomics, 2012
- RACA:Kim et al., PNAS, 2013
- BayesHammer:Nikolenko et al., BMC Genomics, 2013
- SPA:Yang and Yooseph, Nucleic Acids Res., 2013
- SOAPdenovo2:Luo et al., Gigascience, 2012
- JR-Assembler:Chu et al., Proc Natl Acad Sci U S A., 2013
- Platanus:Kajitani et al., Genome Res., 2014
- Omega(メタゲノム用):Haider et al., Bioinformatics, 2014
- MetaVelvet-SL(メタゲノム用):Afiahayati et al., DNA Res., 2015
- MHAP(PacBio用):Berlin et al., Nat Biotechnol., 2015
- Miniasm:Li, H., Bioinformatics, 2016
- ABruijn:Lin et al., Proc Natl Acad Sci U S A, 2016
- HINGE:Kamath et al., Genome Res., 2017
- FastEtch:Ghosh and Kalyanaraman, IEEE/ACM Trans Comput Biol Bioinform., 2019
- Marvel:Nowoshilow et al., Nature, 2018
- TrioCanu (trio binning; ロングリード用):Koren et al., Nat Biotechnol., 2018
- Flye(ロングリード用):Kolmogorov et al., Nat Biotechnol., 2019
- wtdbg2(ロングリード用):Ruan and Li, Nat Methods, 2020
- OPTICALKERMIT(ロングリード用; optical mapping):Leinonen and Salmela, BMC Bioinformatics, 2020
- Shasta(ロングリード用):Shafin et al., Nat Biotechnol., 2020
- HiCanu(ロングリード用):Nurk et al., Genome Res., 2020
- Peregrine(ロングリード用):Chin and Khalak, bioRxiv, 2019
Review、ガイドライン、パイプライン系:
- Review:Miller et al., Genomics, 2010
- CG pipeline(パイプライン; 454用):Kislyuk et al., Bioinformatics, 2010
- A5 pipeline(パイプライン):Tritt et al., PLoS One, 2012
- Review:El-Metwally et al., PLoS Comput Biol., 2013
- iMetAMOS(パイプライン):Koren et al., BMC Bioinformatics, 2014
- MyPro(パイプライン; Prokaryote用):Liao et al., J Microbiol Methods., 2015
- Review:Sohn and Nam, Brief Bioinform., 2018
- ガイドライン系:Dominguez Del Angel et al., F1000Res., 2018
- Review:Senol Cali et al., Brief Bioinform., 2019
- QUAST-LG(評価系):Mikheenko et al., Bioinformatics, 2018
- dnAQET(評価系):Yavas et al., BMC Genomics, 2019
- Review:SoRelle et al., Arch Pathol Lab Med., 2020
- Merqury(評価系):Rhie et al., Genome Biol., 2020
アセンブル | トランスクリプトーム(転写物)用
Rパッケージはおそらくありません。
プログラム:
- MIRA:Chevreux et al., Genome Res., 2004
- Multiple-k:Surget-Groba and Montoya-Burgos, Genome Res., 2010
- Trans-ABySS:Robertson et al., Nat Methods, 2010
- Rnnotator:Martin et al., BMC Genomics, 2010
- Trinity:Grabherr et al., Nat Biotechnol, 2011
- Oases:Schulz et al., Bioinformatics, 2012
- EBARDenovo:Chu et al., Bioinformatics, 2013
- BRANCH:Bao et al., Bioinformatics, 2013
- IDBA-tran:Peng et al., Bioinformatics, 2013
- SOAPdenovo-Trans:Xie et al., Bioinformatics, 2014
- VTBuilder:Archer et al., BMC Bioinformatics, 2014
- Rockhopper2(バクテリア用):Tjaden B, Genome Biol., 2015
- DETONATE(RSEM-EVAL):Li et al., Genome Biol., 2014
- Bridger:Chang et al., Genome Biol., 2015
- IFRAT:Mbandi et al., BMC Bioinformatics, 2015
- SCERNA(主に植物):Honaas et al., PLoS One, 2016
- BinPacker:Liu et al., PLoS Comput Biol., 2016
- TraRECo(Windows/Linux用):Yoon et al., BMC Genomics, 2018
- TransLiG:Liu et al., Genome Biol., 2019
Review、ガイドライン、パイプライン系:
- Review:Martin and Wang, Nat Rev Genet., 2011
- ガイドライン:Haznedaroglu et al., BMC Bioinformatics, 2012
- Review:Góngora-Castillo, Nat Prod Rep., 2013
- ガイドライン:Yang and Smith, BMC Genomics, 2013
- ガイドライン:O'Neil et al., BMC Genomics, 2013
- ガイドライン:Feldmesser et al., BMC Genomics, 2014
- パイプライン(454用):Melicher et al., BMC Genomics, 2014
- パイプライン(組合せ系; SAMP and CDTA):He et al., BMC Genomics, 2015
- 手法比較(Bridgerがよかった):Rana et al., PLoS One, 2016
- ガイドライン系(multiple-k strategyのどのあたりまでk値を試すかに関する議論):Durai and Schulz, Bioinformatics, 2016
- 手法比較(リファレンス配列があってもde novoをやる価値はあるとのこと):Wang and Gribskov, Bioinformatics, 2017
- 手法比較:Hölzer and Marz, Gigascience, 2019
- ガイドライン系(Trimmomatic, Trinity, Bowtie2, BUSCO, TransRate, and so on):Naranpanawa et al., Sci rep., 2020
マッピング | について
リファレンス配列にマッピングを行うプログラム達です。
basic aligner (unspliced aligner)はsplice-aware aligner (spliced aligner)内部で使われていたりします。
R以外(basic aligner; unspliced aligner):
- SSAHA2:Ning et al., Genome Res., 2001
- RMAP:Smith et al., BMC Bioinformatics, 2008
- MAQ:Li et al., Genome Res., 2008
- PASS:Campagna et al., Bioinformatics, 2009
- MOM:Eaves and Gao, Bioinformatics, 2009
- Bowtie:Langmead et al., Genome Biol., 2009
- BWA:Li and Durbin, Bioinformatics, 2009(BWA-shortの論文)
- SHRiMP:Rumble et al., PLoS Comput. Biol., 2009
- SOAP2:Li et al., Bioinformatics, 2009
- RazerS:Weese et al., Genome Res., 2009
- PerM:Chen et al., Bioinformatics, 2009
- BFAST:Homer et al., PLoS One, 2009
- BWA:Li and Durbin, Bioinformatics, 2010(BWA-SWの論文)
- Novoalign:Krawitz et al., Bioinformatics, 2010
- GASSST:Rizk and Lavenier, Bioinformatics, 2010
- Stampy:Lunter and Goodson, Genome Res., 2011
- SHRiMP2:David et al., Bioinformatics, 2011
- BarraCUDA:Klus et al., BMC Res Notes, 2012 (樋口 千洋 氏提供情報)
- SOAP3:Liu et al., Bioinformatics, 2012
- FANSe:Zhang et al., Nucleic Acids Res., 2012
- Bowtie 2:Langmead and Salzberg, Nat. Methods, 2012
- BatMis:Tennakoon et al., Bioinformatics, 2012
- YAHA:Faust and Hall, Bioinformatics, 2012
- CUSHAW2:Liu and Schmidt, Bioinformatics, 2012
- YOABS:Galinsky VL, Bioinformatics, 2012
- CUDASW++ 3.0:Liu et al., BMC Bioinformatics, 2013
- Subread:Liao et al., Nucleic Acids Res., 2013
- SOAP3-dp:Luo et al., PLoS One, 2013
- CUSHAW3:Liu et al., PLoS One, 2014
R以外(splice-aware aligner; spliced aligner):
- BLAT:Kent WJ, Genome Res., 2002seed-and-extend系
- QPALMA:De Bona et al., Bioinformatics, 2008seed-and-extend系
- TopHat:Trapnell et al., Bioinformatics, 2009exon-first系
- RNA-MATE:Cloonan et al., Bioinformatics, 2009
- GSNAP:Wu et al., Bioinformatics, 2010seed-and-extend系
- SpliceMap:Au et al., Nucleic Acids Res., 2010seed-and-extend系
- Supersplat:Bryant et al., Bioinformatics, 2010
- MapSplice:Wang et al., Nucleic Acids Res., 2010multi-seed系
- HMMSplicer:Dimon et al., PLoS One, 2010
- X-MATE:Wood et al., Bioinformatics, 2011
- RUM:Grant et al., Bioinformatics, 2011
- RNASEQR:Chen et al., Nucleic Acids Res., 2012
- PASSion:Zhang et al., Bioinformatics, 2012
- SOAPsplice:Huang et al., Front Genet., 2011
- ContextMap:Bonfert et al., BMC Bioinformatics, 2012
- OSA:Hu et al., Bioinformatics, 2012
- STAR:Dobin et al., Bioinformatics, 2013
- TrueSight:Li et al., Nucleic Acids Res., 2013
- OLego:Wu et al., Nucleic Acids Res., 2013
- TopHat2:Kim et al., Genome Biol., 2013multi-seed系
- segemehl:Hoffmann et al., Genome Biol., 2014
- HSA:Bu et al., BMC Syst Biol., 2013
- FineSplice:Gatto et al., Nucleic Acids Res., 2014
- lack(segemehlの一部):Otto et al., Bioinformatics, 2014
- HISAT:Kim et al., Nat Methods, 2015(高橋 広夫 氏提供情報)
- ALFALFA:Vyverman et al., BMC Bioinformatics, 2015
- HISAT2(HISATとTopHat2の後継?!):原著論文はまだ(高橋 広夫 氏提供情報)
R以外(Bisulfite sequencing (BS-seq)用):
- BSMAP:Xi and Li, BMC Bioinformatics, 2009
- BS Seeker:Chen et al., BMC Bioinformatics, 2010
- Pash 3.0:Coarfa et al., BMC Bioinformatics, 2010
- Bismark:Krueger et al., Bioinformatics, 2011
- B-SOLANA(SOLiD用?!):Kreck et al., Bioinformatics, 2012
- BRAT-BW:Harris et al., Bioinformatics, 2012
- segemehl:Otto et al., Bioinformatics, 2012
- BatMeth:Lim et al., Genome Biol., 2012
- PASS-bis:Campagna et al., Bioinformatics, 2013
- BS-Seeker2:Guo et al., BMC Genomics, 2013
- GNUMAP-bs:Hong et al., BMC Bioinformatics, 2013
マッピング | basic aligner
アルゴリズム的な観点から、Seed-and-extend系とBurrows-Wheeler transform (BWT)系の大きく2種類に大別可能です。
そのアルゴリズムの特性によって得手不得手があるようですのでご注意ください。
また、論文自体は古くても、プログラムを頻繁にバージョンアップさせてよりよくなっている場合もありますので、出版年にこだわる必要はあまりないのではと思っています。
プログラム:
- SSAHA2:Ning et al., Genome Res., 2001
- RMAP:Smith et al., BMC Bioinformatics, 2008
- MAQ:Li et al., Genome Res., 2008
- PASS:Campagna et al., Bioinformatics, 2009
- MOM:Eaves and Gao, Bioinformatics, 2009
- Bowtie:Langmead et al., Genome Biol., 2009
- BWA:Li and Durbin, Bioinformatics, 2009(BWA-shortの論文)
- SHRiMP:Rumble et al., PLoS Comput. Biol., 2009
- SOAP2:Li et al., Bioinformatics, 2009
- RazerS:Weese et al., Genome Res., 2009
- PerM:Chen et al., Bioinformatics, 2009
- BFAST:Homer et al., PLoS One, 2009
- BWA:Li and Durbin, Bioinformatics, 2010(BWA-SWの論文)
- Novoalign:Krawitz et al., Bioinformatics, 2010
- GASSST:Rizk and Lavenier, Bioinformatics, 2010
- Stampy:Lunter and Goodson, Genome Res., 2011
- SHRiMP2:David et al., Bioinformatics, 2011
- BarraCUDA:Klus et al., BMC Res Notes, 2012 (樋口 千洋 氏提供情報)
- SOAP3:Liu et al., Bioinformatics, 2012
- FANSe:Zhang et al., Nucleic Acids Res., 2012
- Bowtie 2:Langmead and Salzberg, Nat. Methods, 2012
- BatMis:Tennakoon et al., Bioinformatics, 2012
- YAHA:Faust and Hall, Bioinformatics, 2012
- CUSHAW2:Liu and Schmidt, Bioinformatics, 2012
- YOABS:Galinsky VL, Bioinformatics, 2012
- CUDASW++ 3.0:Liu et al., BMC Bioinformatics, 2013
- Subread:Liao et al., Nucleic Acids Res., 2013
- SOAP3-dp:Luo et al., PLoS One, 2013
- CUSHAW3:Liu et al., PLoS One, 2014
以下は、2012年頃に書いたものですので情報自体は若干古いですが分類別にリストアップしています。
(BLASTやFASTA的な昔からある)Seed-and-extend methods
このカテゴリに含まれる方法は、マップするshort readよりもさらに短い配列(これがいわゆる"seed"; 種)の完全一致(perfect match)領域をリファレンス配列から探し、このseed領域を拡張(extend)させてアラインメントするというスタンスのものたちです。特徴としては、マップするリファレンス配列(ゲノム配列 or トランスクリプトーム配列)のクオリティが低い場合にも対応可能です。したがって、近縁種のゲノムにマップしたいような場合には、内部的にこのカテゴリに含まれるプログラムを採用しているほうがより多くマップさせることができることが期待されます。また、polymorphism (or sequence variation)の割合が高いようなものについても、こちらのカテゴリの方法のほうがよりよい結果を得られると期待されます。
以下のプログラムたちがこのカテゴリに含まれます:
- SSAHA2:Ning et al., Genome Res., 2001
- RMAP:Smith et al., BMC Bioinformatics, 2008
- MAQ:Li et al., Genome Res., 2008
- MOM:Eaves and Gao, Bioinformatics, 2009
- SHRiMP:Rumble et al., PLoS Comput. Biol., 2009
- RazerS:Weese et al., Genome Res., 2009
- PerM:Chen et al., Bioinformatics, 2009
- BFAST:Homer et al., PLoS One, 2009
- Novoalign:Krawitz et al., Bioinformatics, 2010
- GASSST:Rizk and Lavenier, Bioinformatics, 2010
- Stampy:Lunter and Goodson, Genome Res., 2011
- SHRiMP2:David et al., Bioinformatics, 2011
- FANSe:Zhang et al., Nucleic Acids Res., 2012
Burrows-Wheeler transform (BWT) methods
このカテゴリに含まれる方法は、文字通りBWTというアルゴリズムを用いて効率的にマップしたいshort readsと完全一致(perfect match)する領域を探すものたちです。基本的にperfect matchを探すのに向いているため、ミスマッチを許容すると劇的に計算が遅くなります。したがって、比較的きれいなトランスクリプトーム配列に対してマップさせたい場合には、このカテゴリに含まれるプログラムを用いるほうが計算時間的に有利だと思います。short readのころには、このアルゴリズムを実装したプログラムがどんどんpublishされていたような印象がありますが、最近はどうなんでしょうね。こっち方面はあまりサーベイしていないのですが、プログラムのバージョンアップで多少のindelsに対しても頑健なものになっているのかもしれません。。。
以下のプログラムたちがこのカテゴリに含まれます:
- Bowtie:Langmead et al., Genome Biol., 2009
- BWA:Li and Durbin, Bioinformatics, 2009(BWA-shortの論文)
- SOAP2:Li et al., Bioinformatics, 2009
- BWA:Li and Durbin, Bioinformatics, 2010(BWA-SWの論文)
- Bowtie 2:Langmead and Salzberg, Nat. Methods, 2012
マッピング | splice-aware aligner
basic alignerはジャンクションリード(junciton reads; spliced reads)のマッピングができません。
splice-aware alignerは計算に時間がかかるものの、それらもマッピングしてくれます。
SpliceMapは、リードの半分の長さをマップさせておいてそのアラインメントを拡張(extend)させる戦略を採用しています。
いずれの方法も、バージョンアップがどんどんなされるプログラムは、アルゴリズム(計算手順)自体も変わっていたりしますので参考程度にしてください。
大きな分類としては、seed-and-extend系とexon-first系に分けられるようです。
exon-first系は、内部的にbasic-alignerを用いてざっくりとマップできるものをマップし、マップされなかったものがジャンクションリード候補としてリードを分割してマップされる場所を探すイメージです。
seed-and-extend系は、前者に比べて計算時間がかかるものの、全リードを等価に取り扱うため、exon-first系にありがちな
「本当はジャンクションリードなんだけどbasic alignerでのマッピング時にpseudogeneにマップされてしまう」ということはないようです(Garber et al., 2011)。
その後multi-seed系の方法も提案されているようです(Gatto et al., 2014)。HISAT2というHISATとTopHat2の後継プログラムもあるようです(高橋 広夫 氏提供情報)。
プログラム:
- BLAT:Kent WJ, Genome Res., 2002seed-and-extend系
- QPALMA:De Bona et al., Bioinformatics, 2008seed-and-extend系
- TopHat:Trapnell et al., Bioinformatics, 2009exon-first系
- RNA-MATE:Cloonan et al., Bioinformatics, 2009
- GSNAP:Wu et al., Bioinformatics, 2010seed-and-extend系
- SpliceMap:Au et al., Nucleic Acids Res., 2010seed-and-extend系
- Supersplat:Bryant et al., Bioinformatics, 2010
- MapSplice:Wang et al., Nucleic Acids Res., 2010multi-seed系
- HMMSplicer:Dimon et al., PLoS One, 2010
- X-MATE:Wood et al., Bioinformatics, 2011
- RUM:Grant et al., Bioinformatics, 2011
- RNASEQR:Chen et al., Nucleic Acids Res., 2012
- PASSion:Zhang et al., Bioinformatics, 2012
- SOAPsplice:Huang et al., Front Genet., 2011
- ContextMap:Bonfert et al., BMC Bioinformatics, 2012
- OSA:Hu et al., Bioinformatics, 2012
- STAR:Dobin et al., Bioinformatics, 2013
- TrueSight:Li et al., Nucleic Acids Res., 2013
- OLego:Wu et al., Nucleic Acids Res., 2013
- TopHat2:Kim et al., Genome Biol., 2013multi-seed系
- segemehl:Hoffmann et al., Genome Biol., 2014
- HSA:Bu et al., BMC Syst Biol., 2013
- FineSplice:Gatto et al., Nucleic Acids Res., 2014
- lack(segemehlの一部):Otto et al., Bioinformatics, 2014
- HISAT:Kim et al., Nat Methods, 2015
- ALFALFA:Vyverman et al., BMC Bioinformatics, 2015
- HISAT2(HISATとTopHat2の後継?!):原著論文はまだ
マッピング | Bisulfite sequencing用
Bisulfite sequencingデータ専用のマッピングプログラムも結構あります。
プログラム:
- BSMAP:Xi and Li, BMC Bioinformatics, 2009
- BS Seeker:Chen et al., BMC Bioinformatics, 2010
- Pash 3.0:Coarfa et al., BMC Bioinformatics, 2010
- Bismark:Krueger et al., Bioinformatics, 2011
- B-SOLANA(SOLiD用?!):Kreck et al., Bioinformatics, 2012
- BRAT-BW:Harris et al., Bioinformatics, 2012
- segemehl:Otto et al., Bioinformatics, 2012
- BatMeth:Lim et al., Genome Biol., 2012
- PASS-bis:Campagna et al., Bioinformatics, 2013
- BS-Seeker2:Guo et al., BMC Genomics, 2013
- GNUMAP-bs:Hong et al., BMC Bioinformatics, 2013
マッピング | (ESTレベルの長さの) contig
NGSというよりは一般的なものですが、いくつか挙げておきます。
Trans-ABySSの論文中では、exonerate(のest2genomeというモード)でcontigをマウスゲノムにマップしています(Robertson et al., Nat Methods, 2010)。
また、SPALNはcDNA配列をゲノムにマップするものですが、私自身がEST配列をゲノムにマップする目的で使用した経験があります。
マッピング | 基礎
マッピングの基本形を示します。出力はBED形式と似ています(理解しやすいので...)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
イントロ | 一般 | ランダムな塩基配列を作成の4.を実行して得られたmulti-FASTAファイル(hoge4.fa)です。
in_f1 <- "hoge4.fa"
in_f2 <- "data_reads.fasta"
out_f <- "hoge.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
reads <- readDNAStringSet(in_f2, format="fasta")
out <- c("in_f2", "in_f1", "start", "end")
for(i in 1:length(reads)){
hoge <- vmatchPattern(pattern=as.character(reads[i]), subject=fasta)
hoge1 <- cbind(start(unlist(hoge)), end(unlist(hoge)))
hoge2 <- names(unlist(hoge))
hoge3 <- rep(as.character(reads[i]), length(hoge2))
out <- rbind(out, cbind(hoge3, hoge2, hoge1))
}
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
マッピング | single-end | ゲノム | basic aligner(基礎) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてsingle-end RNA-seqデータのリファレンスゲノム配列へのマッピングを行うやり方を示します。
basic alignerの一つであるBowtie (Langmead et al., Genome Biol., 2009)を実装した
Rbowtieパッケージを内部的に使っています。
入力として与えるRNA-seqデータファイルはFASTA形式でもFASTQ形式でも構いません。ただし、拡張子が".fa", ".fna", ".fasta", ".fq", ".fastq"のいずれかでないといけないようです。
例えば".txt"だとエラーになります。また、圧縮ファイルでも構わないようです。".gz", ".bz2", ".xz"を認識できるようです。
リファレンスゲノムは、BSgenomeパッケージで利用可能なものをそのまま指定することができるようです。
つまり、available.genomes()でみられるパッケージ名を指定可能だということです。もし指定したパッケージがインストールされていなかった場合でも、自動でインストールしてくれるようです。
マッピングプログラム(aligner)のデフォルトは、ジャンクションリードのマッピングができないが高速なBowtie (Langmead et al., Genome Biol., 2009)です。
Bowtieプログラム自体は、複数個所にマップされるリードの取り扱い(uniquely mapped reads or multi-mapped reads)を"-m"オプションで指定したり、
許容するミスマッチ数を指定する"-v"などの様々なオプションを利用可能ですが、QuasR中では、"-m 1 -–best -–strata"オプションを基本として、内部的に自動選択するらしいです。
実際に用いられたオプションは下記スクリプト中のoutオブジェクトの出力結果から知ることができます。
この項目では、マッピングのオプションについては変更を加えずに、一つのRNA-seqファイルのマッピングを行う基本的なやり方を示しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
mapping_single_genome1.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(sample_RNAseq1.fa)、
そして2行目の2列目に「任意のサンプル名」(例:namae)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName"と"SampleName"のままにしておくということです)
マッピング後に得られるBAM形式ファイルは、"sample_RNAseq1_XXXXXXXXXX.bam"というファイル名で作業ディレクトリ上に自動で生成されます。 ここで、XXXXXXXXXXはランダムな文字列からなります。
理由は、同じRNA-seqファイルを異なるパラメータやリファレンス配列にマッピングしたときに、同じ名前だと上書きしてしまう恐れがあるためです。
また、Quality Controlレポートも"sample_RNAseq1_XXXXXXXXXX_QC.pdf"というファイル名で作業ディレクトリ上に自動で生成されます。
マッピングに用いたパラメータは"-m 1 --best --strata -v 2"であったことがわかります。
in_f1 <- "mapping_single_genome1.txt"
in_f2 <- "ref_genome.fa"
library(QuasR)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
mapping_single_genome2.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(sample_RNAseq1.fa.gz)、
そして2行目の2列目に「任意のサンプル名」(例:asshuku)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName"と"SampleName"のままにしておくということです)
圧縮ファイルをそのまま読み込ませることもできる例です。
in_f1 <- "mapping_single_genome2.txt"
in_f2 <- "ref_genome.fa"
library(QuasR)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
mapping_single_genome2.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(sample_RNAseq1.fa.gz)、
そして2行目の2列目に「任意のサンプル名」(例:asshuku)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName"と"SampleName"のままにしておくということです)
マッピング結果をBED形式ファイルとして保存するやり方です。GenomicAlignmentsパッケージを用いて内部的にBAM形式ファイルを読み込み、
BED形式に変換してから保存しています。qQCReport関数実行時に警告メッセージ(compressed 'fasta' input is not yet supported)が出ることを確認していますが、
単純にgzip圧縮FASTA形式ファイルはまだサポートされていないということだけで、マッピング自体はうまくいっているので問題ありません。
in_f1 <- "mapping_single_genome2.txt"
in_f2 <- "ref_genome.fa"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName"と"SampleName"のままにしておくということです)
一つもマップされるものがない例であり、FASTQを入力ファイルとして読み込めるという例でもあります。もちろん圧縮ファイル形式でもOKです。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "hoge4.fa"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
5.サンプルデータ7のFASTQ形式ファイル(SRR037439.fastq)のトキソプラズマゲノムBSgenome.Tgondii.ToxoDB.7.0へのマッピングの場合:
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName"と"SampleName"のままにしておくということです)
ヒトRNA-seqデータをBSgenomeパッケージで利用可能な生物種のリファレンスゲノム配列にマッピングするやり方の一例です。
計算時間短縮のためゲノムサイズの小さいトキソプラズマゲノムBSgenome.Tgondii.ToxoDB.7.0を指定しています。
BSgenome.Hsapiens.UCSC.hg19を指定すると(おそらく数時間程度かかるかもしれませんが...)マップされる確率は当然上がります(同じ生物種なので)。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "BSgenome.Tgondii.ToxoDB.7.0"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マッピング | single-end | ゲノム | basic aligner(応用) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてsingle-end RNA-seqデータのリファレンスゲノム配列へのマッピングを行うやり方を示します。
basic alignerの1つであるBowtie (Langmead et al., Genome Biol., 2009)を実装した
Rbowtieパッケージを内部的に使っています。
Bowtie自体は、複数個所にマップされるリードの取り扱い(uniquely mapped reads or multi-mapped reads)を"-m"オプションで指定したり、
許容するミスマッチ数を指定する"-v"などの様々なオプションを利用可能ですが、「基礎」のところではやり方を示しませんでした。
ここでは、マッピングのオプションをいくつか変更して挙動を確認したり、複数のRNA-seqファイルを一度にマッピングするやり方を示します。
尚、出力ファイルは、"*.bam", "*_QC.pdf", "*.bed"の3つです。それ以外のファイルは基本無視で大丈夫です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
オプションを"-m 1 --best --strata -v 0"とした例です。
sample_RNAseq1.faでマップされないのは計3リードです。
2リード("chr3_11_45"と"chr3_15_49")はchr5にもマップされるので、"-m 1"オプションで落とされます。
1リード("chr5_1_35")は該当箇所と完全一致ではない(4番目の塩基にミスマッチをいれている)ので落とされます。
in_f1 <- "mapping_single_genome1.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-m 1 --best --strata -v 0"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
オプションを"-m 1 --best --strata -v 1"とした例です。
sample_RNAseq1.faでマップされないのは計2リードです。
chr5にもマップされる2リード("chr3_11_45"と"chr3_15_49")が"-m 1"オプションで落とされます。
in_f1 <- "mapping_single_genome1.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-m 1 --best --strata -v 1"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
オプションを"-m 1 -v 1"とした例です。
sample_RNAseq1.faでマップされないのは計3リードです。
2リード("chr3_11_45"と"chr3_15_49")はchr5にもマップされるので、"-m 1"オプションで落とされます。
1リード("chr3_3_37")は該当箇所と完全一致ですが、chr5_3_37とは1塩基ミスマッチでマップ可能です。
"--best --strata"は最小のミスマッチ数でヒットした結果のみ出力するオプションなので、
これをつけておかないと"chr3_3_37"は2か所にマップされるリードということで"-m 1"オプションで落とされる、という例です。
in_f1 <- "mapping_single_genome1.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-m 1 -v 1"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
複数のRNA-seqデータファイルを一度にマッピングするときに用意するファイルの記述例です。
下の行にどんどんマップしたいファイルを追加していくだけです。リード長が異なっていても大丈夫なようです。
sample_RNAseq1.faでマップされないのは計2リードです。
chr5にもマップされる2リード("chr3_11_45"と"chr3_15_49")が"-m 1"オプションで落とされます。
sample_RNAseq2.faでマップされないのは、2-4番目のジャンクションリードです。
in_f1 <- "mapping_single_genome4.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-m 1 --best --strata -v 1"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
5. gzip圧縮FASTQ形式ファイル(SRR609266.fastq.gz)のカイコゲノム(integretedseq.fa)へのマッピングの場合(mapping_single_genome7.txt):
small RNA-seqデータ(400Mb弱、11928428リード)です。
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものが
SRR609266.fastq.gz (Nie et al., BMC Genomics, 2013)です。
カイコゲノム配列は、農業生物資源研究所(NIAS)が提供しているカイコゲノム配列のウェブページからIntegrated sequences (integretedseq.txt.gz)
をダウンロードし、解凍します。解凍後のファイル名は"integretedseq.txt"となりますが、拡張子を".txt"から".fa"に変更して、"integretedseq.fa"としたものを使用しています。
ちなみに、イントロ | 一般 | 配列取得 | ゲノム配列 | 公共DBからを参考にして、
Ensemblから取得したBombyx_mori.GCA_000151625.1.22.dna.toplevel.fa.gzを解凍したものだと、
qAlign関数実行中に「これはfastaファイルではない」とエラーが出て実行できませんでした。20分強かかります。
in_f1 <- "mapping_single_genome7.txt"
in_f2 <- "integretedseq.fa"
param_mapping <- "-m 1 --best --strata -v 1"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
6. 2つのgzip圧縮FASTQ形式ファイル(SRR609266.fastq.gzとhoge4.fastq.gz)のカイコゲノム(integretedseq.fa)へのマッピングの場合(mapping_single_genome8.txt):
small RNA-seqデータ(400Mb弱; 11928428リード; Nie et al., BMC Genomics, 2013)です。
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものがSRR609266.fastq.gzです。
また、前処理 | トリミング | アダプター配列除去(基礎) | ShortRead(Morgan_2009)の4を実行して得られたものがhoge4.fastq.gzです。
カイコゲノム配列は、農業生物資源研究所(NIAS)が提供しているカイコゲノム配列のウェブページ
からIntegrated sequences (integretedseq.txt.gz)
をダウンロードし、解凍します。解凍後のファイル名は"integretedseq.txt"となりますが、拡張子を".txt"から".fa"に変更して、"integretedseq.fa"としたものを使用しています。30分強かかります。
in_f1 <- "mapping_single_genome8.txt"
in_f2 <- "integretedseq.fa"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マップしたいFASTQファイルリストおよびそのサンプル名を記述したsrp017142_samplename.txtを作業ディレクトリに保存したうえで、下記を実行します。
BSgenomeパッケージで利用可能なBSgenome.Hsapiens.UCSC.hg19へマッピングしています。
名前から推測できるように"UCSC"の"hg19"にマップしているのと同じです。
basic alignerの一つであるBowtieを内部的に用いており、
ここではマッピング時のオプションを"-m 1 --best --strata -v 2"にしています。10時間程度かかります。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
7.と基本的に同じですが、parallelパッケージを用いて並列計算するやり方(高橋 広夫 氏提供情報) です。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
library(parallel)
time_s <- proc.time()
cl <- makeCluster(detectCores()/2)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
stopCluster(cl)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マッピング | single-end | ゲノム | splice-aware aligner | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてsingle-end RNA-seqデータのリファレンスゲノム配列へのマッピングを行うやり方を示します。
splice-aware alignerの一つであるSpliceMap (Au et al., Nucleic Acids Res., 2010)を実装した
Rbowtieパッケージを内部的に使っています。
(QuasRパッケージ中の)SpliceMap利用時は、リード長が50bp以上あることが条件のようです。
したがって、35bpのsample_RNAseq1.faを入力ファイルに含めるとエラーが出ます。
尚、出力ファイルは、"*.bam", "*_QC.pdf", "*.bed"の3つです。それ以外のファイルは基本無視で大丈夫です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
デフォルトのオプションで実行する例です。
リード長が50bp以上ありますが、なぜか「failed while generating 25mers」という類のエラーが出ます。
in_f1 <- "mapping_single_genome5.txt"
in_f2 <- "ref_genome.fa"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2,
splicedAlignment=T)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
オプションを"-max_intron 200 -min_intron 5 -max_multi_hit 5 -selectSingleHit TRUE -seed_mismatch 1 -read_mismatch 2 -try_hard yes"とした例です。
リード長が50bp以上ありますが、なぜか「failed while generating 25mers」という類のエラーが出ます。
in_f1 <- "mapping_single_genome5.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-max_intron 200 -min_intron 5 -max_multi_hit 5 -selectSingleHit TRUE -seed_mismatch 1 -read_mismatch 2 -try_hard yes"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2,
alignmentParameter=param_mapping,
splicedAlignment=T)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
オプションを"-max_intron 200 -min_intron 5 -max_multi_hit 5 -selectSingleHit TRUE -seed_mismatch 1 -read_mismatch 2 -try_hard yes"とした例です。
リード長が50bp以上ありますが、なぜか「failed while generating 25mers」という類のエラーが出ます。
in_f1 <- "mapping_single_genome6.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-max_intron 200 -min_intron 5 -max_multi_hit 5 -selectSingleHit TRUE -seed_mismatch 1 -read_mismatch 2 -try_hard yes"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2,
alignmentParameter=param_mapping,
splicedAlignment=T)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
デフォルトのオプションで実行する例です。
実際に用いられたオプションが"-max_intron 400000 -min_intron 20000 -max_multi_hit 10 -selectSingleHit TRUE -seed_mismatch 1 -read_mismatch 2 -try_hard yes"となっているのがわかります。
エラーが出なくなりますが当然?!マップされません。
in_f1 <- "mapping_single_genome6.txt"
in_f2 <- "ref_genome.fa"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2,
splicedAlignment=T)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
オプションを"-max_intron 400000 -min_intron 20000 -max_multi_hit 10 -selectSingleHit TRUE -seed_mismatch 1 -read_mismatch 2 -try_hard yes"とした例です。
4.と同じ結果(エラーは出ないがマップされない)になります。
in_f1 <- "mapping_single_genome6.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-max_intron 400000 -min_intron 20000 -max_multi_hit 10 -selectSingleHit TRUE -seed_mismatch 1 -read_mismatch 2 -try_hard yes"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2,
alignmentParameter=param_mapping,
splicedAlignment=T)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マッピング | paired-end | ゲノム | basic aligner(基礎) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてpaired-end RNA-seqデータのリファレンスゲノム配列へのマッピングを行うやり方を示します。
basic alignerの1つであるBowtie (Langmead et al., Genome Biol., 2009)を実装した
Rbowtieパッケージを内部的に使っています。
mapping_paired_genome1.txtのような2行目の1列目と2列目に「マッピングしたいRNA-seqファイル名1と2」
(例:sample_RNAseq_1.faとsample_RNAseq_2.fa)、
そして2行目の3列目に「任意のサンプル名」(例:namae)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName1", "FileName2",および"SampleName"のままにしておくということです)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
乳酸菌RNA-seqデータSRR616268の最初の100万リード分です。
SRR616268sub_1.fastq.gzは、74,906,576 bytes、全リード107 bpです。
SRR616268sub_2.fastq.gzは、67,158,462 bytes、全リード93 bpです。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)
がリファレンス配列です。マッピングオプションはデフォルトです。
in_f1 <- "mapping_paired_genome1.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
1.の入力ファイルから5'および3'側をrcode_20150707_preprocessing.txtに書いてある手順でトリム
して得られた998,521リードからなるpaired-endのファイルです。
SRR616268sub_trim3_1.fastq.gz (59,092,219 bytes)と
SRR616268sub_trim3_2.fastq.gz (54,667,920 bytes)です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)
がリファレンス配列です。マッピングオプションはデフォルトです。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マッピング | paired-end | ゲノム | basic aligner(応用) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてpaired-end RNA-seqデータのリファレンスゲノム配列へのマッピングを行うやり方を示します。
basic alignerの1つであるBowtie (Langmead et al., Genome Biol., 2009)を実装した
Rbowtieパッケージを内部的に使っています。
Bowtie自体は、複数個所にマップされるリードの取り扱い(uniquely mapped reads or multi-mapped reads)を"-m"オプションで指定したり、
許容するミスマッチ数を指定する"-v"などの様々なオプションを利用可能ですが、「基礎」のところではやり方を示しませんでした。
ここでは、マッピングのオプションをいくつか変更して挙動を確認したり、複数のRNA-seqファイルを一度にマッピングするやり方を示します。
尚、出力ファイルは、"*.bam", "*_QC.pdf", "*.bed"の3つです。それ以外のファイルは基本無視で大丈夫です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
998,521リードからなるpaired-endのファイルです。
SRR616268sub_trim3_1.fastq.gz (59,092,219 bytes)と
SRR616268sub_trim3_2.fastq.gz (54,667,920 bytes)です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)
がリファレンス配列です。
オプションを"-m 1 --best --strata -v 0"とした例です。
-m 1で1か所にのみマップされるリード、
-v 0で許容するミスマッチ数を0個にしています。
--best --strataは、許容するミスマッチ数が1以上の場合に効果を発揮します。
ここでは意味をなしませんが、つけておいて悪さをするものではないので、通常は無条件でつけます。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
param_mapping <- "-m 1 --best --strata -v 0"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
1.と基本的に同じで、オプションを"-m 1 --best --strata -v 2"とした例です。
-m 1で1か所にのみマップされるリード、
-v 2で許容するミスマッチ数を2個にしています。
--best --strataをつけておくことで、例えば同じリードがミスマッチ数0個(perfect match)でマップされるのが1か所のみだったにもかかわらず、
ミスマッチ数1 or 2個でもマップされるされる別の場所があったときに-m 1の効果で複数個所にマップされるとして出力されなくなる不幸をなくすことができます。
もっともよい層(best strata)のみで評価するという捉え方でいいでしょう。これがどうしても嫌だというヒトは普通はいませんので、通常は無条件でつけます。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マッピング | paired-end | トランスクリプトーム | basic aligner(基礎) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてpaired-end RNA-seqデータのリファレンスゲノム配列へのマッピングを行うやり方を示します。
basic alignerの1つであるBowtie (Langmead et al., Genome Biol., 2009)を実装した
Rbowtieパッケージを内部的に使っています。
mapping_paired_genome1.txtのような2行目の1列目と2列目に「マッピングしたいRNA-seqファイル名1と2」
(例:sample_RNAseq_1.faとsample_RNAseq_2.fa)、
そして2行目の3列目に「任意のサンプル名」(例:namae)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName1", "FileName2",および"SampleName"のままにしておくということです)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
乳酸菌RNA-seqデータSRR616268の998,521リードからなるpaired-endのファイルです。
SRR616268sub_trim3_1.fastq.gz (59,092,219 bytes)と
SRR616268sub_trim3_2.fastq.gz (54,667,920 bytes)です。
リファレンス配列は「イントロ | 一般 | 配列取得 | トランスクリプトーム配列 | GenomicFeatures(Lawrence_2013)」
の例題6を実行して得られたもの(transcriptome_Lcasei12A.fasta)を利用します。
マッピングオプションはデフォルトです。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "transcriptome_Lcasei12A.fasta"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マッピング | paired-end | トランスクリプトーム | basic aligner(応用) | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてpaired-end RNA-seqデータのリファレンスゲノム配列へのマッピングを行うやり方を示します。
basic alignerの1つであるBowtie (Langmead et al., Genome Biol., 2009)を実装した
Rbowtieパッケージを内部的に使っています。
mapping_paired_genome1.txtのような2行目の1列目と2列目に「マッピングしたいRNA-seqファイル名1と2」
(例:sample_RNAseq_1.faとsample_RNAseq_2.fa)、
そして2行目の3列目に「任意のサンプル名」(例:namae)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName1", "FileName2",および"SampleName"のままにしておくということです)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
乳酸菌RNA-seqデータSRR616268の998,521リードからなるpaired-endのファイルです。
SRR616268sub_trim3_1.fastq.gz (59,092,219 bytes)と
SRR616268sub_trim3_2.fastq.gz (54,667,920 bytes)です。
リファレンス配列は「イントロ | 一般 | 配列取得 | トランスクリプトーム配列 | GenomicFeatures(Lawrence_2013)」
の例題6を実行して得られたもの(transcriptome_Lcasei12A.fasta)を利用します。
オプションを"-m 1 --best --strata -v 0"とした例です。
-m 1で1か所にのみマップされるリード、
-v 0で許容するミスマッチ数を0個にしています。
--best --strataは、許容するミスマッチ数が1以上の場合に効果を発揮します。
ここでは意味をなしませんが、つけておいて悪さをするものではないので、通常は無条件でつけます。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "transcriptome_Lcasei12A.fasta"
param_mapping <- "-m 1 --best --strata -v 0"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マップ後 | について
(ゲノムやトランスクリプトーム配列へのマッピング時には問題にならないと思いますが、おそらく)RNA-seqのアセンブルを行う場合には、sequencing errorの除去以外にも「アダプター配列」や「low-complexity reads (低複雑性のリード;繰り返し配列)」や「PCR duplicates(ロングインサートライブラリによくあるらしい...)」の除去を行うのが普通なようです。(私がTrinityでアセンブルを行う場合にはそんなことやったこともないのですが、やったら大分違うのでしょうか。。。)ここではこれらのerror除去を行うプログラムを列挙しておきます:
入力ファイルがリファレンス配列へのマップ後のファイル(SAM/BAM, BED, GFF形式など)(Rパッケージ):
入力ファイルがリファレンス配列へのマップ後のファイル(SAM/BAM, BED, GFF形式など)(R以外):
(おそらく)454 platform用:
その他:
- SHREC: Schroder et al., Bioinformatics, 2009; 入力ファイル形式がよくわからなかったのでそれ以上深入りせず...
- TagDust: Lassmann et al., Bioinformatics, 2009; web上でマニュアルを読めなかったのでそれ以上深入りせず...
- SeqTrim: Falqueras et al., BMC Bioinformatics, 2010; loginを要求されたのでそれ以上深入りせず...
- CUDA-EC: Shi et al., J. Comput. Biol., 2010; web上でマニュアルを読めなかったのでそれ以上深入りせず...
- EDAR: Zhao et al., J. Comput. Biol., 2010
- Redeem: Yang et al., BMC Bioinformatics, 2011; documentationが不親切でよくわかりません...orz
- PBcR: Koren et al., Nat Biotechnol., 2012; Celera Assemblerの一部として実装されているらしい
マップ後 | 出力ファイル形式について
様々ないろいろな出力ファイル形式があることがわかります。
注目すべきは、Sequence Alignment/Map (SAM) formatです。この形式は国際共同研究の1000人のゲノムを解析するという1000 Genomes Projectで採用された(開発された)フォーマットで、("@"から始まる)header sectionと(そうでない)alignment sectionから構成されています。このヒトの目で解読可能な形式がSAMフォーマットで、このバイナリ版がBinary Alignment/Map (BAM)フォーマットというものです。今後SAM/BAM formatという記述をよく見かけるようになることでしょう。
代表的な出力ファイル形式:
マップ後 | 出力ファイルの読み込み | BAM形式 | について
マッピング結果の標準出力ファイルは、SAM/BAM形式と呼ばれるファイルです(Li et al., Bioinformatics, 2009)。
2016年頃からは、PacBioの生の出力ファイルもBAM形式になっています。
ここではSAM/BAM形式ファイルを読み込めるものをリストアップします。
マップ後 | 出力ファイルの読み込み | BAM形式 | rbamtools(Kaisers_2015)
rbamtoolsパッケージを用いてBAM形式ファイルを読み込むやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample_RNAseq1.bam"
library(rbamtools)
hoge <- bamReader(in_f)
hoge
マップ後 | 出力ファイルの読み込み | BAM形式 | GenomicAlignments(Lawrence_2013)
GenomicAlignmentsパッケージを用いてBAM形式ファイルを読み込むやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample_RNAseq1.bam"
library(GenomicAlignments)
hoge <- readGAlignments(in_f)
hoge
マップ後 | 出力ファイルの読み込み | Bowtie形式
Bowtie形式出力ファイルを読み込むやり方を、yeastRNASeqパッケージ中にある(このパッケージがインストールされていることが前提です)wt_1_f.bowtie.gzファイルを例として解説します。yeastRNASeqというパッケージがインストールされていれば、(例えば私のWindows 7, 64bitマシン環境だと)「コンピュータ - OS(C:) - Program Files - R - R-3.0.1 - library - yeastRNASeq - reads」から目的のファイルにたどり着きます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. ディレクトリの変更をちゃんとやった場合:
in_f <- "wt_1_f.bowtie.gz"
library(ShortRead)
aligned <- readAligned(in_f, type="Bowtie")
aligned
2. yeastRNASeqパッケージ中のreadsフォルダ中に目的のファイルがあることが既知の場合:
in_f <- "wt_1_f.bowtie.gz"
param1 <- "yeastRNASeq"
param2 <- "reads"
library(ShortRead)
path <- file.path(system.file(package=param1, param2))
aligned <- readAligned(path, in_f, type="Bowtie")
aligned
マップ後 | 出力ファイルの読み込み | SOAP形式
basic alignerの一つであるSOAPを実行して得られたSOAP形式ファイルの読み込み例をいくつか示します。
SOAP以外のフォーマットも「マッピング | 出力ファイルの読み込み |」を参考にして、必要な情報さえ読み込んでおけば、それ以降は同じです。
SOAP形式ファイルの場合、param1に相当するのは入力ファイルの8列目の記述に、そしてparam2に相当するのは7列目の記述に相当します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
885リードからなるファイルです。8番染色体上の+鎖のみにマップされたリードのみFASTAファイルで保存するやり方1です。
in_f <- "output.soap"
out_f <- "hoge1.fasta"
param1 <- "chr08"
param2 <- "+"
library(ShortRead)
aligned <- readAligned(in_f, type="SOAP")
aligned
aligned <- aligned[aligned@chromosome == param1]
aligned
aligned <- aligned[aligned@strand == "+"]
aligned
fasta <- sread(aligned)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
885リードからなるファイルです。8番染色体上の+鎖のみにマップされたリードのみFASTAファイルで保存するやり方2です。
in_f <- "output.soap"
out_f <- "hoge2.fasta"
param1 <- "chr08"
param2 <- "+"
library(ShortRead)
aligned <- readAligned(in_f, type="SOAP")
aligned
hoge <- compose(chromosomeFilter(param1), strandFilter(param2))
aligned <- aligned[hoge(aligned)]
fasta <- sread(aligned)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
885リードからなるファイルです。strandは気にせずに10番染色体上にマップされたリードのみFASTAファイルで保存するやり方です。
in_f <- "output.soap"
out_f <- "hoge3.fasta"
param1 <- "chr10"
library(ShortRead)
aligned <- readAligned(in_f, type="SOAP")
aligned
hoge <- compose(chromosomeFilter(param1))
aligned <- aligned[hoge(aligned)]
fasta <- sread(aligned)
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
染色体ごとにマップされたリード数情報を取得するやり方です。
in_f <- "output.soap"
out_f <- "hoge4.txt"
library(ShortRead)
aligned <- readAligned(in_f, type="SOAP")
aligned
out <- as.data.frame(table(aligned@chromosome))
colnames(out) <- c("chromosome", "frequency")
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
染色体およびstrandごとにマップされたリード数情報を取得するやり方です。
in_f <- "output.soap"
out_f <- "hoge5.txt"
library(ShortRead)
aligned <- readAligned(in_f, type="SOAP")
hoge <- paste(aligned@chromosome, aligned@strand, sep="___")
out <- as.data.frame(table(hoge))
colnames(out) <- c("chromosome___strand", "frequency")
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
マップ後 | 出力ファイルの読み込み | htSeqTools (Planet_2012)
比較解析などの場合に「複数のサンプル間のクラスタリングをまず行う」ことをトランスクリプトーム解析の基本手順として行ったほうがよい、ということを私の講義でも述べていますが、このパッケージでも1) サンプル間の類似度をMDSプロットで表現する機能があるようですね。
また、2) PCR biasに起因する一部のリードが大量にシークエンスされてしまったようなものも、BIC (Bayesian Information Criterion)を用いたリピート数のモデル化ややFDR (False Discovery Rate)の閾値を設けて取り除くようなこともできるようです。
ChIP-seq (or MeDIP, DNase-seq)などの場合には、controlに対してimmuno-precipitation (IP)サンプルにおける特定の領域中のリード数(coverage)が増大します。このcoverageの標準偏差(standard deviation; SD)はリード数nのルートに比例するらしいです。なのでもしIPサンプルのSDn (=SD/sqrt(n))がcontrolサンプルのSDnと同程度であればIPがうまくいっていないことを意味します。このパッケージでは、この3) enrichment efficiencyの評価もできるようです。
その他には、ゲノムにマップされた領域のスクリーニングや、領域ごとにマップされたリード数をカウントしたり、複数サンプル間でのリード数の比較をlikelihod ratioやpermutationカイ二乗検定で行ってくれるようです。
ちなみに、入力ファイルの形式はBAM形式ファイル(Rsamtools or ShortReadパッケージで読み込み可能)かBED, GFF, WIG形式ファイル(rtracklayerパッケージで読み込み可能)で、マップ後のファイルということになります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- ""
library(htSeqTools)
マップ後 | カウント情報取得 | について
BAM形式などのマッピング結果ファイルからカウントデータを取得するためのパッケージや関数もいくつかあります。
htSeqToolsのislandCounts関数、
Rsubread(Windows版はなし)のfeatureCounts関数(Liao et al., Bioinformatics, 2014)、
GenomicRangesのsummarizeOverlaps関数などです。
QuasRは、内部的にGenomicRangesのsummarizeOverlaps関数を利用しています。
scaterは、内部的にkallisto
(Bray et al., Nat Biotechnol., 2016)を利用しており、トランスクリプトーム配列へのマッピングからカウント情報取得まで行ってくれるようです。
Rパッケージ:
- htSeqTools:Planet et al., Bioinformatics., 2012
- BitSeq(getExpression関数。入力はSAMファイル):Glaus et al., Bioinformatics, 2012
- easyRNASeq:Delhomme et al., Bioinformatics, 2012
- Rsubread:Liao et al., Nucleic Acids Res., 2013
- GenomicRanges:Lawrence et al., PLoS Comput. Biol., 2013
- QuasR:Gaidatzis et al., Bioinformatics, 2015
- scater (scRNA-seq用):McCarthy et al., Bioinformatics, 2017
R以外:
- BEDTools(coverageBedプログラム;BAMをBEDに変換する手間はある):Quinlan and Hall, Bioinformatics, 2010
- HTSeq:Anders et al., Bioinformatics, 2015
- Rcount:Schmid and Grossniklaus, Bioinformatics, 2015
- kallisto(マッピングからカウント情報まで): Bray et al., Nat Biotechnol., 2016
- Salmon(マッピングからカウント情報まで): Patro et al., Nat Methods, 2017
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)
QuasRパッケージを用いたsingle-end RNA-seqデータのリファレンスゲノム配列へのBowtieによるマッピングから、
カウントデータ取得までの一連の流れを示します。アノテーション情報は、GenomicFeatures
パッケージ中の関数を利用してTxDbオブジェクトをネットワーク経由で取得するのを基本としつつ、
TxDbパッケージを読み込むやり方も示しています。
マッピングのやり方やオプションの詳細については
マッピング | single-end | ゲノム | basic aligner(応用) | QuasR(Gaidatzis_2015)などを参考にしてください。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で(オプションを"-m 1 --best --strata -v 2"として)行っています。
hg19にマップした結果なので、TxDbオブジェクト取得時のゲノム情報もそれを基本として、
UCSC Genes ("knownGene")を指定しているので、Entrez Gene IDに対するカウントデータ取得になっています。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f <- "hoge1.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param_txdb1 <- "hg19"
param_txdb2 <- "knownGene"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
txdb <- makeTxDbFromUCSC(genome=param_txdb1, tablename=param_txdb2)
txdb
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
tmp <- cbind(rownames(count), count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で
(オプションを"-m 1 --best --strata -v 2"として)行っています。
hg19にマップした結果なので、TxDbオブジェクト取得時のゲノム情報もそれを基本として、
RefSeq Genes ("refGene")を指定しているのでRefSeq Gene IDに対するカウントデータ取得になっていますはずですが、そうはなっていません。
この理由は(バグか開発者のポリシーかは不明ですが)おそらくRefSeqが転写物単位のIDなためゲノムマッピング結果の取り扱いとは相性が悪いためでしょう。
ちなみに長さ情報は変わっているので、なんらかの形でRefSeqの情報が反映されているのでしょう。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f <- "hoge2.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param_txdb1 <- "hg19"
param_txdb2 <- "refGene"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
txdb <- makeTxDbFromUCSC(genome=param_txdb1, tablename=param_txdb2)
txdb
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
tmp <- cbind(rownames(count), count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で
(オプションを"-m 1 --best --strata -v 2"として)行っています。
hg19にマップした結果なので、TxDbオブジェクト取得時のゲノム情報もそれを基本として
Ensembl Genes ("ensGene")を指定しているので、Ensembl Gene IDに対するカウントデータ取得になっています。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f <- "hoge3.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param_txdb1 <- "hg19"
param_txdb2 <- "ensGene"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
txdb <- makeTxDbFromUCSC(genome=param_txdb1, tablename=param_txdb2)
txdb
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
tmp <- cbind(rownames(count), count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で
(オプションを"-m 1 --best --strata -v 2"として)行っています。
hg19にマップした結果なので、TxDbオブジェクト取得時のゲノム情報もそれを基本として
Ensembl Genes ("ensGene")を指定しているので、Ensembl Gene IDに対するカウントデータ取得になっています。
基本は3と同じですが、一般的なカウントデータ行列の形式(2列目以降がカウント情報)にし、配列長情報と別々のファイルにして保存するやり方です。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f1 <- "hoge4_count.txt"
out_f2 <- "hoge4_genelength.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param_txdb1 <- "hg19"
param_txdb2 <- "ensGene"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
txdb <- makeTxDbFromUCSC(genome=param_txdb1, tablename=param_txdb2)
txdb
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で
(オプションを"-m 1 --best --strata -v 2"として)行っています。
3.と違って、TxDbオブジェクト取得時に、Ensemblにアクセスしてhsapiens_gene_ensembl
をデータベース名として指定しています。
ちなみにこれは、txdbオブジェクトの作成まではうまくいきますが、最後のqCount関数の実行のところでエラーが出ます。
理由は、Ensemblで提供されている染色体名seqnames(transcripts(txdb))の中にBSgenome.Hsapiens.UCSC.hg19で提供されている染色体名以外の名前のものが含まれているためです。
データ提供元が異なるとうまくいかない例です。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f <- "hoge5.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param_txdb_ds <- "hsapiens_gene_ensembl"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
txdb <- makeTxDbFromBiomart(dataset=param_txdb_ds)
txdb
count <- qCount(out, txdb, reportLevel=param_reportlevel)
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
マップしたいFASTQファイルリストおよびそのサンプル名を記述したsrp017142_samplename.txtを作業ディレクトリに保存したうえで、下記を実行します。
BSgenome.Hsapiens.UCSC.hg19へマッピングしています。
名前から推測できるように"UCSC"の"hg19"にマップしているのと同じです。
basic alignerの一つであるBowtieを内部的に用いており、
ここではマッピング時のオプションを"-m 1 --best --strata -v 2"にしています。
ここでの目的はUCSC known genesのgene-levelのカウントデータを得ることです。10時間程度かかります。
TxDbオブジェクトのバージョン次第で行数やカウント数は若干変化しますがhoge6.txtのような感じになっていれば正解です。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f <- "hoge6.txt"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
param_mapping <- "-m 1 --best --strata -v 2"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicAlignments)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
count <- qCount(out, txdb, reportLevel=param_reportlevel)
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
6と基本的に同じです。UCSC known genesのgene-levelとexon-levelの両方のカウントデータを同時に得るやり方です。
TxDbオブジェクトのバージョン次第で行数やカウント数は若干変化しますがhoge7_count_gene.txtや
hoge7_count_exon.txtのような感じになっていれば正解です。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f1 <- "hoge7_count_gene.txt"
out_f2 <- "hoge7_count_exon.txt"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
count <- qCount(out, txdb, reportLevel="gene")
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
data_gene <- data
count <- qCount(out, txdb, reportLevel="exon")
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
data_exon <- data
7と基本的に同じです。Quality Control (QC)レポートファイルも作成するやり方です。
TxDbオブジェクトのバージョン次第で行数やカウント数は若干変化しますがhoge8_QC.pdf、
hoge8_count_gene.txtや
hoge8_count_exon.txtのような感じになっていれば正解です。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f1 <- "hoge8_count_gene.txt"
out_f2 <- "hoge8_count_exon.txt"
out_f3 <- "hoge8_QC.pdf"
param_txdb <- "TxDb.Hsapiens.UCSC.hg19.knownGene"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
qQCReport(out, pdfFilename=out_f3)
library(param_txdb, character.only=T)
tmp <- ls(paste("package", param_txdb, sep=":"))
txdb <- eval(parse(text=tmp))
count <- qCount(out, txdb, reportLevel="gene")
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
data_gene <- data
count <- qCount(out, txdb, reportLevel="exon")
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
data_exon <- data
4と8を組み合わせたようなやり方です。数値だけのUCSC gene IDではなくEnsembl Gene IDで取り扱うやり方です。
TxDbオブジェクトのバージョン次第で行数やカウント数は若干変化しますがhoge9_QC.pdf、
hoge9_count_gene.txtや
hoge9_count_exon.txtのような感じになっていれば正解です。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f1 <- "hoge9_count_gene.txt"
out_f2 <- "hoge9_count_exon.txt"
out_f3 <- "hoge9_QC.pdf"
param_txdb1 <- "hg19"
param_txdb2 <- "ensGene"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicFeatures)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
qQCReport(out, pdfFilename=out_f3)
txdb <- makeTxDbFromUCSC(genome=param_txdb1, tablename=param_txdb2)
txdb
count <- qCount(out, txdb, reportLevel="gene")
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
data_gene <- data
count <- qCount(out, txdb, reportLevel="exon")
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
data_exon <- data
マップする側のファイルは、サンプルデータ47のFASTA形式ファイル(sample_RNAseq4.fa)です。
マップされる側のファイルは、Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa)
です。マッピング結果に対して、GFF3形式のアノテーションファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3)
を読み込んでカウント情報を取得しています。
in_f1 <- "mapping_single_genome7.txt"
in_f2 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"
in_f3 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3"
out_f <- "hoge10.txt"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f3, format="auto")
txdb
out <- qAlign(in_f1, in_f2)
alignmentStats(out)
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
tmp <- cbind(rownames(count), count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題10と基本的に同じですが、カウント情報を取得するqCount関数の実行時に、shift = 1オプションを付けています。
in_f1 <- "mapping_single_genome7.txt"
in_f2 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa"
in_f3 <- "Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3"
out_f <- "hoge11.txt"
param_reportlevel <- "gene"
param_shift <- 1
library(QuasR)
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f3, format="auto")
txdb
out <- qAlign(in_f1, in_f2)
alignmentStats(out)
count <- qCount(out, txdb, reportLevel=param_reportlevel,
shift=param_shift)
dim(count)
head(count)
tmp <- cbind(rownames(count), count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | HTSeq(Anders_2015)
HTSeqというPythonプログラムを用いてカウント情報を得るやり方を示します。
ここでは、「マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)」
の例題10を実行して得られたマッピング結果(sample_RNAseq4_3b6c652a602a.bam)を利用します。
これは、Bowtieをデフォルトオプションで実行したものです。
マップする側のファイルは、サンプルデータ47のFASTA形式ファイル(sample_RNAseq4.fa)です。
マップされる側のファイルは、Ensembl Bacteriaから提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.dna.chromosome.Chromosome.fa)
です。
対応するGFF3形式のアノテーションファイルはLactobacillus_hokkaidonensis_jcm_18461.GCA_000829395.1.30.chromosome.Chromosome.gff3ですが、
ファイル名が長いと見づらいので、hoge.gff3として取り扱います。
対応するGTF形式のアノテーションファイル(hoge1.gtf)は、「イントロ | ファイル形式の変換 | GFF3 --> GTF」の例題1で作成したものです。
また、sample_RNAseq4_3b6c652a602a.bamも長いので、hoge.bamとして取り扱います。
1. GFF3でgeneレベルのカウントデータを取得する場合:
アノテーションファイルがGFF3形式であるという前提です。
hoge.gff3の3列目のgeneでレベル指定、9列目のgene_idでfeature IDを指定
(gene_idの代わりにIDでもOK)しています
。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GFF3_gene.txtです。2,194 genesですね。
htseq-count -t gene -i gene_id -f bam hoge.bam hoge.gff3 > output_GFF3_gene.txt
2. GFF3でtranscriptレベルのカウントデータを取得する場合:
アノテーションファイルがGFF3形式であるという前提です。
hoge.gff3の3列目のtranscriptでレベル指定、9列目のtranscript_idでfeature IDを指定
(transcript_idの代わりにIDやParentでもOK)しています。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GFF3_transcript.txtです。2,250 transcriptsですね。
htseq-count -t transcript -i transcript_id -f bam hoge.bam hoge.gff3 > output_GFF3_transcript.txt
3. GFF3でexonレベルのカウントデータを取得する場合:
アノテーションファイルがGFF3形式であるという前提です。
hoge.gff3の3列目のexonでレベル指定、9列目のexon_idでfeature IDを指定
(exon_idの代わりにParentやNameでもOK)しています。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GFF3_exon.txtです。2,262 exonsですね。
htseq-count -t exon -i exon_id -f bam hoge.bam hoge.gff3 > output_GFF3_exon.txt
4. GFF3でCDSレベルのカウントデータを取得する場合:
アノテーションファイルがGFF3形式であるという前提です。
hoge.gff3の3列目のCDSでレベル指定、9列目のprotein_idでfeature IDを指定
(protein_idの代わりにIDやParentでもOK)しています。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GFF3_CDS.txtです。2,194 CDSsですね。
htseq-count -t CDS -i protein_id -f bam hoge.bam hoge.gff3 > output_GFF3_CDS.txt
5. GTFでgeneレベルのカウントデータを取得する場合:
アノテーションファイルがGTF形式であるという前提です。
hoge1.gtfの3列目のgeneでレベル指定、9列目のgene_idでfeature IDを指定
(gene_idの代わりにIDでもOK)しています。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GTF_gene.txtです。2,194 genesですね。
htseq-count -t gene -i gene_id -f bam hoge.bam hoge1.gtf > output_GTF_gene.txt
6. GTFでtranscriptレベルのカウントデータを取得する場合:
アノテーションファイルがGTF形式であるという前提です。
hoge1.gtfの3列目のtranscriptでレベル指定、9列目のtranscript_idでfeature IDを指定
(transcript_idの代わりにIDやParentでもOK)しています。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GTF_transcript.txtです。2,250 transcriptsですね。
htseq-count -t transcript -i transcript_id -f bam hoge.bam hoge1.gtf > output_GTF_transcript.txt
7. GTFでexonレベルのカウントデータを取得する場合:
アノテーションファイルがGTF形式であるという前提です。
hoge1.gtfの3列目のexonでレベル指定、9列目のexon_idでfeature IDを指定
(exon_idの代わりにParentでもOK)しています。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GTF_exon.txtです。2,262 exonsですね。
htseq-count -t exon -i exon_id -f bam hoge.bam hoge1.gtf > output_GTF_exon.txt
8. GTFでCDSレベルのカウントデータを取得する場合:
アノテーションファイルがGTF形式であるという前提です。
hoge1.gtfの3列目のCDSでレベル指定、9列目のIDでfeature IDを指定
(IDの代わりにprotein_idやParentでもOKだが、protein_idだとちょっと変)しています。
マッピング結果がBAMファイル(hoge.bam)なので-f bamとしています(SAMの場合はsam)。
出力ファイルはoutput_GTF_CDS.txtです。2,194 CDSsですね。
htseq-count -t CDS -i ID -f bam hoge.bam hoge1.gtf > output_GTF_CDS.txt
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション無 | QuasR(Gaidatzis_2015)
QuasRパッケージを用いたsingle-end RNA-seqデータのリファレンスゲノム配列へのBowtieによるマッピングから、
カウントデータ取得までの一連の流れを示します。アノテーション情報がない場合を想定しているので、GenomicAlignments
パッケージを利用して、マップされたリードの和集合領域(union range)を得たのち、領域ごとにマップされたリード数をカウントしています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
オプションを"-m 1 --best --strata -v 0"とした例です。
in_f1 <- "mapping_single_genome1.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-m 1 --best --strata -v 0"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
tmpfname <- out@alignments[,1]
tmpsname <- out@alignments[,2]
for(i in 1:length(tmpfname)){
if(i == 1){
k <- readGAlignments(tmpfname[i])
} else{
k <- c(k, readGAlignments(tmpfname[i]))
}
}
m <- reduce(granges(k))
tmp <- as.data.frame(m)
for(i in 1:length(tmpfname)){
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(tmp, count)
}
out_f <- sub(".bam", "_range.txt", tmpfname[i])
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
2.サンプルデータ7のFASTQ形式ファイル(SRR037439.fastq)の
BSgenomeパッケージで利用可能なBSgenome.Hsapiens.UCSC.hg19
へのマッピング結果の場合:
mapping_single_genome3.txtのような2行目の1列目に「マッピングしたいRNA-seqファイル名」(SRR037439.fastq)、
そして2行目の2列目に「任意のサンプル名」(例:human_brain)を記載したタブ区切りテキストファイルを用意した上で(オプションを"-m 1 --best --strata -v 2"として)行っています。
RNA-seqデータのほうのリード数は少ないですが、リファレンス配列の前処理でかなり時間がかかるようです(2時間とか...)。
in_f1 <- "mapping_single_genome3.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
alignmentStats(out)
tmpfname <- out@alignments[,1]
tmpsname <- out@alignments[,2]
for(i in 1:length(tmpfname)){
k <- readGAlignments(tmpfname[i])
m <- reduce(granges(k))
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(as.data.frame(m), count)
out_f <- sub(".bam", "_range.txt", tmpfname[i])
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
}
複数のRNA-seqデータファイルごとに和集合領域を定めてカウント情報を得るやり方です。"*_range.txt"という2つの出力ファイルが生成されます。
in_f1 <- "mapping_single_genome4.txt"
in_f2 <- "ref_genome.fa"
param_mapping <- "-m 1 --best --strata -v 1"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
tmpfname <- out@alignments[,1]
tmpsname <- out@alignments[,2]
for(i in 1:length(tmpfname)){
k <- readGAlignments(tmpfname[i])
m <- reduce(granges(k))
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(as.data.frame(m), count)
out_f <- sub(".bam", "_range.txt", tmpfname[i])
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
}
全部のマッピング結果をまとめて和集合領域を定め、カウント情報を得るやり方です。サンプル間比較の際に便利。
in_f1 <- "mapping_single_genome4.txt"
in_f2 <- "ref_genome.fa"
out_f <- "hoge4.txt"
param_mapping <- "-m 1 --best --strata -v 1"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
tmpfname <- out@alignments[,1]
tmpsname <- out@alignments[,2]
for(i in 1:length(tmpfname)){
if(i == 1){
k <- readGAlignments(tmpfname[i])
} else{
k <- c(k, readGAlignments(tmpfname[i]))
}
}
m <- reduce(granges(k))
tmp <- as.data.frame(m)
for(i in 1:length(tmpfname)){
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(tmp, count)
}
out_f <- sub(".bam", "_range.txt", tmpfname[i])
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
全部のマッピング結果をまとめて和集合領域を定め、カウント情報を得るやり方です。一般的なカウントデータ行列の形式(2列目以降がカウント情報)にし、配列長情報と別々のファイルにして保存するやり方です。
in_f1 <- "mapping_single_genome4.txt"
in_f2 <- "ref_genome.fa"
out_f1 <- "hoge5_count.txt"
out_f2 <- "hoge5_genelength.txt"
param_mapping <- "-m 1 --best --strata -v 1"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
tmpfname <- out@alignments[,1]
tmpsname <- out@alignments[,2]
for(i in 1:length(tmpfname)){
if(i == 1){
k <- readGAlignments(tmpfname[i])
} else{
k <- c(k, readGAlignments(tmpfname[i]))
}
}
m <- reduce(granges(k))
h <- as.data.frame(m)
tmp <- paste(h[,1],h[,2],h[,3],h[,4],h[,5], sep="_")
for(i in 1:length(tmpfname)){
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(tmp, count)
}
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- paste(h[,1],h[,2],h[,3],h[,4],h[,5], sep="_")
tmp <- cbind(tmp, h$width)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F, col.names=T)
マップ後 | カウント情報取得 | paired-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)
QuasRパッケージを用いたpaired-end RNA-seqデータ
のリファレンスゲノム配列へのBowtieによるマッピングから、
カウントデータ取得までの一連の流れを示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
乳酸菌RNA-seqデータSRR616268の最初の100万リード分のpaired-endファイル(SRR616268sub_1.fastq.gzと
SRR616268sub_2.fastq.gz)から5'および3'側を
rcode_20150707_preprocessing.txtに書いてある手順でトリム
して得られた998,521リードからなるpaired-endのファイルです。
SRR616268sub_trim3_1.fastq.gz (59,092,219 bytes)と
SRR616268sub_trim3_2.fastq.gz (54,667,920 bytes)です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)
とGFF3形式のアノテーションファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3)
を読み込むやり方です。マッピングオプションはデフォルトです。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f3 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f <- "hoge1.txt"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f3, format="auto")
txdb
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
tmp <- cbind(rownames(count), count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
1.の出力ファイルは2列目が配列長、3列目がカウント情報でしたが、これを2つのファイルに分割して出力させるやり方を示します。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f3 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f1 <- "hoge2_count.txt"
out_f2 <- "hoge2_length.txt"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
txdb <- makeTxDbFromGFF(in_f3, format="auto")
txdb
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
data <- as.data.frame(count[,-1])
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
2.と基本的に同じですが、BEDファイルも出力させています。GenomicAlignmentsパッケージが提供するreadGAlignments関数を利用しています。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f3 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f1 <- "hoge3_count.txt"
out_f2 <- "hoge3_length.txt"
param_reportlevel <- "gene"
library(QuasR)
library(GenomicFeatures)
library(GenomicAlignments)
txdb <- makeTxDbFromGFF(in_f3, format="auto")
txdb
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
data <- as.data.frame(count[,-1])
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
3.と基本的に同じですが、マッピング時のオプションを"-m 1 --best --strata -v 0"とした例です。
-m 1で1か所にのみマップされるリード、
-v 0で許容するミスマッチ数を0個にしています。
--best --strataは、許容するミスマッチ数が1以上の場合に効果を発揮します。
ここでは意味をなしませんが、つけておいて悪さをするものではないので、通常は無条件でつけます。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f3 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f1 <- "hoge4_count.txt"
out_f2 <- "hoge4_length.txt"
param_reportlevel <- "gene"
param_mapping <- "-m 1 --best --strata -v 0"
library(QuasR)
library(GenomicFeatures)
library(GenomicAlignments)
txdb <- makeTxDbFromGFF(in_f3, format="auto")
txdb
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
data <- as.data.frame(count[,-1])
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
4.と基本的に同じですが、マッピング時のオプションを"-m 1 --best --strata -v 2"とした例です。
-v 2で許容するミスマッチ数を2個にしているので、マップされるリード数やカウントの総和が増えるはずです。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
in_f3 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.chromosome.Chromosome.gff3"
out_f1 <- "hoge5_count.txt"
out_f2 <- "hoge5_length.txt"
param_reportlevel <- "gene"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicFeatures)
library(GenomicAlignments)
txdb <- makeTxDbFromGFF(in_f3, format="auto")
txdb
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
count <- qCount(out, txdb, reportLevel=param_reportlevel)
dim(count)
head(count)
data <- as.data.frame(count[,-1])
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マップ後 | カウント情報取得 | paired-end | ゲノム | アノテーション無 | QuasR(Gaidatzis_2015)
QuasRパッケージを用いたpaired-end RNA-seqデータのリファレンスゲノム配列へのBowtieによるマッピングから、
カウントデータ取得までの一連の流れを示します。アノテーション情報がない場合を想定しているので、GenomicAlignments
パッケージを利用して、マップされたリードの和集合領域(union range)を得たのち、領域ごとにマップされたリード数をカウントしています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
乳酸菌RNA-seqデータSRR616268の最初の100万リード分のpaired-endファイル(SRR616268sub_1.fastq.gzと
SRR616268sub_2.fastq.gz)から5'および3'側を
rcode_20150707_preprocessing.txtに書いてある手順でトリム
して得られた998,521リードからなるpaired-endのファイルです。
SRR616268sub_trim3_1.fastq.gz (59,092,219 bytes)と
SRR616268sub_trim3_2.fastq.gz (54,667,920 bytes)です。
Ensembl (Zerbino et al., Nucleic Acids Res., 2018)から提供されている
Lactobacillus casei 12Aの
multi-FASTA形式ゲノム配列ファイル(Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa)
がリファレンス配列です。マッピングオプションはデフォルトです。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "Lactobacillus_casei_12a.GCA_000309565.2.25.dna.chromosome.Chromosome.fa"
out_f1 <- "hoge1_count.txt"
out_f2 <- "hoge1_genelength.txt"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
tmpfname <- out@alignments[,1]
tmpsname <- out@alignments[,2]
for(i in 1:length(tmpfname)){
if(i == 1){
k <- readGAlignments(tmpfname[i])
} else{
k <- c(k, readGAlignments(tmpfname[i]))
}
}
m <- reduce(granges(k))
h <- as.data.frame(m)
tmp <- paste(h[,1],h[,2],h[,3],h[,4],h[,5], sep="_")
for(i in 1:length(tmpfname)){
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(tmp, count)
}
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- paste(h[,1],h[,2],h[,3],h[,4],h[,5], sep="_")
tmp <- cbind(tmp, h$width)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F, col.names=T)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
マップ後 | カウント情報取得 | paired-end | トランスクリプトーム | QuasR(Gaidatzis_2015)
QuasRパッケージを用いてpaired-end RNA-seqデータのリファレンストランスクリプトーム配列へのマッピングを行うやり方を示します。
basic alignerの1つであるBowtie (Langmead et al., Genome Biol., 2009)を実装した
Rbowtieパッケージを内部的に使っています。
mapping_paired_genome1.txtのような2行目の1列目と2列目に「マッピングしたいRNA-seqファイル名1と2」
(例:sample_RNAseq_1.faとsample_RNAseq_2.fa)、
そして2行目の3列目に「任意のサンプル名」(例:namae)を記載したタブ区切りテキストファイルを用意した上で行います。
1行目の文字列は変えてはいけません(つまり"FileName1", "FileName2",および"SampleName"のままにしておくということです)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
乳酸菌RNA-seqデータSRR616268の998,521リードからなるpaired-endのファイルです。
SRR616268sub_trim3_1.fastq.gz (59,092,219 bytes)と
SRR616268sub_trim3_2.fastq.gz (54,667,920 bytes)です。
リファレンス配列は「イントロ | 一般 | 配列取得 | トランスクリプトーム配列 | GenomicFeatures(Lawrence_2013)」
の例題6を実行して得られたもの(transcriptome_Lcasei12A.fasta)を利用します。
マッピングオプションはデフォルトです。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "transcriptome_Lcasei12A.fasta"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
1.と基本的に同じですが、マッピング時のオプションを"-m 1 --best --strata -v 0"とした例です。
-m 1で1か所にのみマップされるリード、
-v 0で許容するミスマッチ数を0個にしています。
--best --strataは、許容するミスマッチ数が1以上の場合に効果を発揮します。
ここでは意味をなしませんが、つけておいて悪さをするものではないので、通常は無条件でつけます。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "transcriptome_Lcasei12A.fasta"
out_f1 <- "hoge2_count.txt"
out_f2 <- "hoge2_length.txt"
param_mapping <- "-m 1 --best --strata -v 0"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
tmp_name <- seqnames(seqinfo(FaFile(in_f2)))
tmp_length <- seqlengths(seqinfo(FaFile(in_f2)))
tmp <- cbind(tmp_name, tmp_length)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
tmp <- tmp_name
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
count <- rep(0, length(tmp_name))
names(count) <- tmp_name
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
hoge2 <- hoge[, c("seqnames")]
count[names(table(hoge2))] <- table(hoge2)
tmp <- cbind(tmp, count)
}
colnames(tmp) <- c("name", out@alignments[,2])
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
2.と基本的に同じですが、マッピング時のオプションを"-m 1 --best --strata -v 2"とした例です。
-v 2で許容するミスマッチ数を2個にしているので、マップされるリード数やカウントの総和が増えるはずです。
in_f1 <- "mapping_paired_genome2.txt"
in_f2 <- "transcriptome_Lcasei12A.fasta"
out_f1 <- "hoge3_count.txt"
out_f2 <- "hoge3_length.txt"
param_mapping <- "-m 1 --best --strata -v 2"
library(QuasR)
library(GenomicAlignments)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
time_e - time_s
out
alignmentStats(out)
out_f <- sub(".bam", "_QC.pdf", out@alignments[,1])
qQCReport(out, pdfFilename=out_f)
out_f
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
tmp <- hoge[, c("seqnames","start","end")]
out_f <- sub(".bam", ".bed", tmpfname[i])
out_f
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
}
tmp_name <- seqnames(seqinfo(FaFile(in_f2)))
tmp_length <- seqlengths(seqinfo(FaFile(in_f2)))
tmp <- cbind(tmp_name, tmp_length)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
tmp <- tmp_name
tmpfname <- out@alignments[,1]
for(i in 1:length(tmpfname)){
count <- rep(0, length(tmp_name))
names(count) <- tmp_name
hoge <- readGAlignments(tmpfname[i])
hoge <- as.data.frame(hoge)
hoge2 <- hoge[, c("seqnames")]
count[names(table(hoge2))] <- table(hoge2)
tmp <- cbind(tmp, count)
}
colnames(tmp) <- c("name", out@alignments[,2])
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
マップ後 | カウント情報取得 | トランスクリプトーム | BED形式ファイルから
BED形式のマップ後のファイルと(マップされる側の)リファレンス配列ファイルを読み込んでリファレンス配列の並びで配列長とカウント情報を取得するやり方を示します。
出力ファイルの並びは「リファレンス配列のdescription行」「配列長」「カウント情報」です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. マップされる側の配列(hoge4.fa)中のIDの並びでBED形式ファイル(sample_1.bed)中の出現回数をカウントする場合:
in_f1 <- "sample_1.bed"
in_f2 <- "hoge4.fa"
out_f <- "hoge1.txt"
library(ShortRead)
data <- read.table(in_f1, sep="\t")
fasta <- readDNAStringSet(in_f2, format="fasta")
fasta
head(data)
hoge <- table(data[,1])
head(hoge)
out <- rep(0, length(fasta))
names(out) <- names(fasta)
out[names(hoge)] <- hoge
head(out)
tmp <- cbind(names(out), width(fasta), out)
colnames(tmp) <- c("ID", "Length", "Count")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
2. マップされる側の配列(hoge4.fa)中のIDをソートした並びでBED形式ファイル(sample_1.bed)中の出現回数をカウントする場合:
in_f1 <- "sample_1.bed"
in_f2 <- "hoge4.fa"
out_f <- "hoge2.txt"
library(ShortRead)
data <- read.table(in_f1, sep="\t")
fasta <- readDNAStringSet(in_f2, format="fasta")
fasta
head(data)
hoge <- table(data[,1])
head(hoge)
out <- rep(0, length(fasta))
names(out) <- sort(names(fasta))
out[names(hoge)] <- hoge
head(out)
tmp <- cbind(names(out), width(fasta[names(out)]), out)
colnames(tmp) <- c("ID", "Length", "Count")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
in_f1 <- "SRR002324_t.bed"
in_f2 <- "h_rna.fasta"
out_f <- "hoge3.txt"
library(ShortRead)
data <- read.table(in_f1, sep="\t")
fasta <- readDNAStringSet(in_f2, format="fasta")
fasta
head(data)
hoge <- table(data[,1])
head(hoge)
out <- rep(0, length(fasta))
names(out) <- names(fasta)
out[names(hoge)] <- hoge
head(out)
tmp <- cbind(names(out), width(fasta), out)
colnames(tmp) <- c("ID", "Length", "Count")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
4. マップされる側の配列(h_rna.fasta)中のIDをソートした並びでBED形式ファイル(SRR002324_t.bed)中の出現回数をカウントする場合:
in_f1 <- "SRR002324_t.bed"
in_f2 <- "h_rna.fasta"
out_f <- "hoge4.txt"
library(ShortRead)
data <- read.table(in_f1, sep="\t")
fasta <- readDNAStringSet(in_f2, format="fasta")
fasta
head(data)
hoge <- table(data[,1])
head(hoge)
out <- rep(0, length(fasta))
names(out) <- sort(names(fasta))
out[names(hoge)] <- hoge
head(out)
tmp <- cbind(names(out), width(fasta[names(out)]), out)
colnames(tmp) <- c("ID", "Length", "Count")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
カウント情報取得 | リアルデータ | について
ここではSAM/BAMなどのマッピング結果ファイルからのカウント情報取得ではなく、最初からカウント情報になっているもののありかや、
それらを提供しているデータベースから取得するプログラムを示します。
カウント情報取得 | リアルデータ | SRP061240 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP061240(Yuan et al., Sci Rep., 2016;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
2016年の原著論文では、100 colon cancer (大腸がん)、36 prostate cancer (膀胱がん)、6 pancreatic cancer (膵臓がん)、
50 healthy individuals (健常者)の計192サンプルから得られたplasma extracellular vesicles (血漿中の細胞外小胞)の発現データを取得しています。
例題4までで得られるのは計384サンプルの発現データです。これは各サンプルにつき2つのtechnical replicatesがあるためです。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP061240という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP061240で検索し、
gene列のRSE v2をダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP061240"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×384 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txt(約46MB)です。
out_f <- "hoge2.txt"
param_ID <- "SRP061240"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でSRP061240で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約24MB)を読み込んで、
カウントの数値行列情報のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルの中身はhoge2.txtと同じです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じですが、サンプル名をcolData(rse)$titleで見えているものに変更するやり方です。
理由はここにHealthy Controlが"N"で計50サンプル, Colorectal Cancerが1-4期("1S", "2S", "3S", and "4S")のステージごとに25サンプルずつの計100サンプル、
Pancreatic Cancerが"Pan"で計6サンプル、Prostate Cancerが"PC"で計36サンプルです。種類ごとにソートされてはいません。
サンプルごとに2 technical replicatesがあるので計384サンプル。
出力ファイルはhoge4.txt(58,037 genes×384 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge4.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
colnames(data) <- colData(rse)$title
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題4と基本的に同じですが、サンプル名中に共通して含まれる"Sample_"を除去しています。
出力ファイルはhoge5.txt(58,037 genes×384 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge5.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
hoge <- colData(rse)$title
hoge2 <- gsub("Sample_", "", hoge)
colnames(data) <- hoge2
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題5と基本的に同じですが、サンプル名でソートしているので、結果的にグループごとにソートされます。
出力ファイルはhoge6.txt(58,037 genes×384 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge6.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
hoge <- colData(rse)$title
hoge2 <- gsub("Sample_", "", hoge)
colnames(data) <- hoge2
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題6の発展形として、technical replicatesのデータをマージしています。
出力ファイルはhoge7.txt(58,037 genes×192 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge7.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
hoge <- colData(rse)$title
hoge2 <- gsub("Sample_", "", hoge)
colnames(data) <- hoge2
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
unique_sample <- unique(colnames(data))
hoge <- NULL
for(i in 1:length(unique_sample)){
posi <- which(colnames(data) == unique_sample[i])
if(length(posi) > 1){
hoge <- cbind(hoge, apply(data[, posi], 1, "sum", na.rm=TRUE))
} else {
hoge <- cbind(hoge, data[, posi])
}
}
colnames(hoge) <- unique_sample
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP056295 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP056295(Lavallée et al., Nat Genet., 2015;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
2015年の原著論文では、計415サンプルの急性骨髄性白血病(acute myeloid leukemia; AML)の発現データを取得しており、
そこで示されている生データのID情報は計3つ(
GSE62190,
GSE66917, and
GSE67039)ですが、
ここでダウンロードするSRP056295のデータはGSE67039に相当します。
原著論文では、31 MLL-F AML samplesと384 controls (non–MLL-F AML samples)で、実際31 vs. 384で2群間比較もなされているようです
SRP056295のオリジナルは525サンプルのようですが、例題4までで得られるのは計520サンプルの発現データです。
これは、カウントデータ取得時にrecount内でのクオリティ基準を満たさなかったため、5サンプル分(SRR1918519, SRR1918416, SRR1918413, SRR1918382, and SRR1918259)が落とされているからです。
520サンプルから同一サンプルの重複除去を行うと、263サンプルになります。
31 MLL-F AML samplesの情報は、原著論文中のSupplementary Table 1にあります。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP056295という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP056295で検索し、
gene列のRSE v2を
ダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP056295"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×520 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txt(約80MB)です。
out_f <- "hoge2.txt"
param_ID <- "SRP056295"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でSRP056295で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約71MB)を読み込んで、
カウントの数値行列情報のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルの中身はhoge2.txtと同じです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じですが、サンプル名をcolData(rse)$titleで見えているものに変更するやり方です。
出力ファイルはhoge4.txt(58,037 genes×520 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge4.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
colnames(data) <- colData(rse)$title
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題4の発展形として、technical replicatesのデータをマージしています。
出力ファイルはhoge5.txt(58,037 genes×263 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge5.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
colnames(data) <- colData(rse)$title
colnames(data)
unique_sample <- unique(colnames(data))
hoge <- NULL
for(i in 1:length(unique_sample)){
posi <- which(colnames(data) == unique_sample[i])
if(length(posi) > 1){
hoge <- cbind(hoge, apply(data[, posi], 1, "sum", na.rm=TRUE))
} else {
hoge <- cbind(hoge, data[, posi])
}
}
colnames(hoge) <- unique_sample
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP056146 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP056146(Costa-Silva et al., Nat Cell Biol., 2015;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
2015年の原著論文では、クッパー細胞(Kupffer cells; 肝臓を構成するの微小組織の1つで、マクロファージの一種)に
92の乾癬(psoriatic)サンプルと82の正常の皮膚サンプルからなる、計12サンプルの発現データを取得しています。
が、例題6までで得られるのは計178サンプルの発現データです。これには5つのtechnical replicatesが含まれているためです(SRS544523, SRS544524, SRS544536, SRS544653, and SRS544667)。
これらをマージすると計173 samplesになります(92 lesional psoriatic skin samples and 81 normal skin samples)。原著論文に比べて1サンプル少ないのは、
SRS544686がカウントデータ取得時にrecount内でのクオリティ基準を満たさなかったため落とされているからです。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
カウント情報取得 | リアルデータ | SRP035988 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP035988(Li et al., J Invest Dermatol., 2014;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
2014年の原著論文では、92の乾癬(psoriatic)サンプルと82の正常の皮膚サンプルからなる、計174サンプルの発現データを取得しています。
が、例題6までで得られるのは計178サンプルの発現データです。これには5つのtechnical replicatesが含まれているためです(SRS544523, SRS544524, SRS544536, SRS544653, and SRS544667)。
これらをマージすると計173 samplesになります(92 lesional psoriatic skin samples and 81 normal skin samples)。原著論文に比べて1サンプル少ないのは、
SRS544686がカウントデータ取得時にrecount内でのクオリティ基準を満たさなかったため落とされているからです。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP035988という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP035988で検索し、
gene列のRSE v2をダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP035988"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×178 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txt(約29MB)です。
out_f <- "hoge2.txt"
param_ID <- "SRP035988"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でSRP035988で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約24MB)を読み込んで、
カウントの数値行列情報のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルの中身はhoge2.txtと同じです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じですが、サンプル名をcolData(rse)$characteristicsで見えているものに変更するやり方です。
理由はここに"psoriatic" or "normal"が分かる情報が含まれているからです。
colData(rse)$characteristicsがリスト形式になっており、colnames(data)に直接代入することができないので、as.character関数をつけています。
出力ファイルはhoge4.txt(58,037 genes×178 samples)です。サンプル名がやたらと長いのが難点。
in_f <- "rse_gene.Rdata"
out_f <- "hoge4.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
colnames(data) <- as.character(colData(rse)$characteristics)
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題4と基本的に同じですが、サンプル名の文字列をスペースで区切り、3番目の要素を抽出しています。
そこが"lesional(病変という意味)" or "normal"のどちらかになるので、必要最小限になるからです。
出力ファイルはhoge5.txt(58,037 genes×178 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge5.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
hoge <- as.character(colData(rse)$characteristics)
hoge2 <- strsplit(hoge, " ", fixed=TRUE)
hoge3 <- unlist(lapply(hoge2, "[[", 3))
colnames(data) <- hoge3
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題5の発展形です。例題5は計178サンプルの並びがグループごとになっていないのでソートしています。
つまり、95 lesional psoriatic samplesと82 normal samplesに分けて出力しています。
出力ファイルはhoge6.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge6.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
hoge <- as.character(colData(rse)$characteristics)
hoge2 <- strsplit(hoge, " ", fixed=TRUE)
hoge3 <- unlist(lapply(hoge2, "[[", 3))
colnames(data) <- hoge3
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
length(grep("lesi", colnames(data)))
length(grep("norma", colnames(data)))
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
technicla replicatesのデータをマージして、
計173 samplesのデータ(92 lesional psoriatic skin samples and 81 normal skin samples)を群ごとにソートしています。
出力ファイルはhoge6.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge7.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
x <- assays(rse)$counts
x <- as.data.frame(x)
data <- cbind(x$SRR1146077, x$SRR1146078, x$SRR1146079,
x$SRR1146080, x$SRR1146081, x$SRR1146082,
x$SRR1146083 + x$SRR1146084, x$SRR1146087, x$SRR1146089,
x$SRR1146090, x$SRR1146091, x$SRR1146092,
x$SRR1146093, x$SRR1146094, x$SRR1146095,
x$SRR1146097, x$SRR1146098 + x$SRR1146099, x$SRR1146100,
x$SRR1146102, x$SRR1146103, x$SRR1146104,
x$SRR1146105, x$SRR1146110, x$SRR1146112,
x$SRR1146119, x$SRR1146123, x$SRR1146124,
x$SRR1146128, x$SRR1146129, x$SRR1146132,
x$SRR1146133, x$SRR1146134, x$SRR1146135,
x$SRR1146136, x$SRR1146140, x$SRR1146141,
x$SRR1146147, x$SRR1146148, x$SRR1146149,
x$SRR1146150, x$SRR1146151, x$SRR1146152,
x$SRR1146154, x$SRR1146155, x$SRR1146156,
x$SRR1146157, x$SRR1146158, x$SRR1146159,
x$SRR1146160, x$SRR1146161, x$SRR1146162,
x$SRR1146163, x$SRR1146164, x$SRR1146165,
x$SRR1146168, x$SRR1146199, x$SRR1146202,
x$SRR1146203, x$SRR1146204, x$SRR1146205,
x$SRR1146206, x$SRR1146208, x$SRR1146209,
x$SRR1146210, x$SRR1146211, x$SRR1146212,
x$SRR1146214, x$SRR1146215, x$SRR1146216 + x$SRR1146217,
x$SRR1146218, x$SRR1146219, x$SRR1146220,
x$SRR1146221, x$SRR1146222, x$SRR1146223,
x$SRR1146224, x$SRR1146225, x$SRR1146226,
x$SRR1146227, x$SRR1146228, x$SRR1146229,
x$SRR1146233, x$SRR1146234, x$SRR1146235,
x$SRR1146237, x$SRR1146239, x$SRR1146240,
x$SRR1146241, x$SRR1146242, x$SRR1146252,
x$SRR1146253, x$SRR1146254,
x$SRR1146076, x$SRR1146085 + x$SRR1146086, x$SRR1146088,
x$SRR1146096, x$SRR1146101, x$SRR1146106,
x$SRR1146107, x$SRR1146108, x$SRR1146109,
x$SRR1146111, x$SRR1146113, x$SRR1146114,
x$SRR1146115, x$SRR1146116, x$SRR1146117,
x$SRR1146118, x$SRR1146120, x$SRR1146121,
x$SRR1146122, x$SRR1146125, x$SRR1146126,
x$SRR1146127, x$SRR1146130, x$SRR1146131,
x$SRR1146137, x$SRR1146138, x$SRR1146139,
x$SRR1146142, x$SRR1146143, x$SRR1146144,
x$SRR1146145, x$SRR1146146, x$SRR1146153,
x$SRR1146166, x$SRR1146167, x$SRR1146169,
x$SRR1146170, x$SRR1146171, x$SRR1146172,
x$SRR1146173, x$SRR1146174, x$SRR1146175,
x$SRR1146176, x$SRR1146177, x$SRR1146178,
x$SRR1146179, x$SRR1146180, x$SRR1146181,
x$SRR1146182, x$SRR1146183, x$SRR1146184,
x$SRR1146185, x$SRR1146186, x$SRR1146187,
x$SRR1146188, x$SRR1146189, x$SRR1146190,
x$SRR1146191, x$SRR1146192, x$SRR1146193,
x$SRR1146194, x$SRR1146195, x$SRR1146196,
x$SRR1146197, x$SRR1146198, x$SRR1146200,
x$SRR1146201, x$SRR1146207, x$SRR1146213,
x$SRR1146230, x$SRR1146231 + x$SRR1146232, x$SRR1146236,
x$SRR1146238, x$SRR1146243, x$SRR1146244,
x$SRR1146245, x$SRR1146246, x$SRR1146247,
x$SRR1146248, x$SRR1146249, x$SRR1146250)
colnames(data) <- c(paste("lesional", 1:92, sep=""), paste("normal", 1:81, sep=""))
rownames(data) <- rownames(x)
dim(data)
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP026126 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP026126(Nottingham et al., RNA, 2016;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
2016年の原著論文で示されている生データのID情報は、
GSE48035とSRP066009です。
ここでダウンロードするSRP026126のデータはGSE48035に相当します。
SRP066009は、原著論文中でも述べられていますがTGIRT-seqのデータ(計12 samples)に相当します。
SRP056295のオリジナルは422サンプルのようですが、例題4までで得られるのは計418サンプルの発現データです。
これは、カウントデータ取得時にrecount内でのクオリティ基準を満たさなかったため、4サンプル分(SRR902979, SRR903120, SRR903123, and SRR903269)が落とされているからです。
418サンプルから同一サンプルの重複除去を行うと、xxxサンプルになります。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP026126という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP026126で検索し、
gene列のRSE v2を
ダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP026126"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×418 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txt(約xxMB)です。
out_f <- "hoge2.txt"
param_ID <- "SRP026126"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でSRP026126で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約68MB)を読み込んで、
カウントの数値行列情報のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルの中身はhoge2.txtと同じです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じですが、サンプル名をcolData(rse)$titleで見えているものに変更するやり方です。
出力ファイルはhoge4.txt(58,037 genes×418 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge4.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
colnames(data) <- colData(rse)$title
colnames(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題4の発展形として、technical replicatesのデータをマージしています。
出力ファイルはhoge5.txt(58,037 genes×62 samples)です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge5.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
colnames(data)
identical(colnames(data), rownames(colData(rse)))
colnames(data) <- colData(rse)$title
colnames(data)
unique_sample <- unique(colnames(data))
hoge <- NULL
for(i in 1:length(unique_sample)){
posi <- which(colnames(data) == unique_sample[i])
if(length(posi) > 1){
hoge <- cbind(hoge, apply(data[, posi], 1, "sum", na.rm=TRUE))
} else {
hoge <- cbind(hoge, data[, posi])
}
}
colnames(hoge) <- unique_sample
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP018853 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP018853(Zhang et al., J Autoimmun., 2016;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
原著論文では、38 healthy samplesと42 high risk of type 1 diabetes (pre-T1D) samplesからなる、計80サンプルのmiRNA発現データを取得しています。
このうち、SRR764979とSRR765019がカウントデータ取得時にrecount内でのクオリティ基準を満たさなかったようです。
結果として、37 healthy samplesと41 pre-T1D samplesの、計78サンプルからなるカウントデータがここでは得られます。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP018853という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP018853で検索し、
gene列のRSE v2をダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP018853"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×78 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txt(約10MB)です。
out_f <- "hoge2.txt"
param_ID <- "SRP018853"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でSRP018853で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約2MB)を読み込んで、
カウントの数値行列情報のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルの中身はhoge2.txtと同じです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じですが、サンプル名をcolData(rse)$characteristicsで見えているものに変更するやり方です。
出力ファイルはhoge4.txt(58,037 genes×78 samples)です。
内訳は、37 healthy samplesと41 pre-T1D samplesです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge4.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
identical(colnames(data), rownames(colData(rse)))
hoge <- colData(rse)$characteristics
hoge2 <- unlist(lapply(hoge, "[[", 1))
hoge3 <- gsub("status: ", "", hoge2)
colnames(data) <- hoge3
colnames(data)
length(grep("heal", colnames(data)))
length(grep("pre-", colnames(data)))
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP012167 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP012167(Haglund et al., J Clin Endocrinol Metab., 2012;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP012167という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP012167で検索し、
gene列のRSE v2をダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP012167"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×25 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txtです。
out_f <- "hoge2.txt"
param_ID <- "SRP012167"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でSRP012167で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約4MB)を読み込んで、
カウントの数値行列情報のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルはhoge3.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP012167 | parathyroidSE(Haglund_2012)
parathyroidSEパッケージを用いて、
SRP012167(Haglund et al., J Clin Endocrinol Metab., 2012;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentオブジェクトという形式のデータセット(parathyroidGenesSEおよびparathyroidExonsSEという名前で格納されています)
をdata関数を用いてロードしたり、カウントデータの数値行列にした状態で保存するやり方を示します。
1. parathyroidGenesSEの場合:
gene-levelカウントデータのRangedSummarizedExperimentオブジェクトです。
63,193 Ensembl gene IDs×27 samplesのカウントデータです。
library(parathyroidSE)
data(parathyroidGenesSE)
ls()
rse <- parathyroidGenesSE
rse
2. parathyroidExonsSEの場合:
exon-levelカウントデータのRangedSummarizedExperimentオブジェクトです。
626,686 exons×27 samplesのカウントデータです。
library(parathyroidSE)
data(parathyroidExonsSE)
ls()
rse <- parathyroidExonsSE
rse
カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP001558(Blekhman et al., Genome Res., 2010;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、カウントデータの数値行列にした状態で保存するやり方を示します。
原著論文では、3生物種(ヒト12 samples、チンパンジー12 samples、そしてアカゲザル12 samples)のカウントデータを取得しています。
ウェブサイトrecount2上でSRP001558で検索すると、
number of samplesが12、speciesがhumanとなっていることから、提供されているカウントデータはhumanに限定されていることがわかります。
例題2までで、なぜか11 samples分のデータしかないことに気づきます。これは、ウェブサイトrecount2上でSRP001558で検索し、
phenotype列のlinkをダウンロードして得られるSRP001558.tsv
を眺めることでなんとなくの理由がわかります。私は、「SRR032117のデータがおかしなことになっており、recount2で提供するクオリティに達しなかった。
このため、recount2のウェブページ上は12 samplesとなっているものの、カウントデータ自体は11 samples分となっているのだろう。」と予想しました。
また、PRJNA119135・GSE17274
・SRA010277はENA上にリンク先がありますが、
ウェブサイトrecount2上では引っかかってきませんでした。
2018年8月7日に、recountWorkflow
で推奨されているscale_counts関数実行後のカウントデータとなるように変更しました。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP001558という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP001558で検索し、
gene列のRSE v2をダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP001558"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×11 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txtです。
out_f <- "hoge2.txt"
param_ID <- "SRP001558"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でSRP001558で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約3MB)を読み込んで、
カウントの数値行列情報(58,037 genes×11 samples)のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルはhoge3.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3とベースとして、さらにサンプルのメタデータ情報ファイル(srp001558_meta_samples.txt)と、
遺伝子(features)のメタデータ情報ファイル(srp001558_meta_features.txt)も出力するやり方です。
58,037 genes×11 samplesからなるカウントデータファイル(hoge4_counts.txt)は列名をSRR...からSRS...に変更しています。
このデータセットの場合は、なぜかtechnical replicatesのサンプルに対して別々のSRS IDが付与されているので、列名変更はほぼ無意味です。
in_f <- "rse_gene.Rdata"
out_f1 <- "hoge4_counts.txt"
out_f2 <- "srp001558_meta_samples.txt"
out_f3 <- "srp001558_meta_features.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
colnames(data) <- colData(rse)$sample
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- colData(rse)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
tmp <- rowData(rse)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
例題4で得られたサンプルのメタデータ情報ファイル(hoge4_meta_samples.txt)中のtitle列に相当する情報で置き換えています。
これは、hoge4_meta_samples.txtをExcelで眺めたときに、たまたまtitle列情報がdiscriminable(容易に識別可能である)だと主観的に判断したためです。
このあたりの情報のクオリティというかどのような情報が提供されているかは、submitter依存です。したがって、一筋縄ではいきません。まるで有益な情報のない残念なものも結構あるからです。
58,037 genes×11 samplesからなる出力ファイルはhoge5.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge5.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
colnames(data)<- colData(rse)$title
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題5の続きのようなものですが、technical replicatesのデータをマージした結果を出力しています。
例えば、"Human female 2 rep1"列と"Human female 2 rep2"列のカウント数の和をとり、列名を"HSF2"のようにしています。
この列名の表記法は、「サンプルデータ42の20,689 genes×18 samplesのリアルカウントデータ
(sample_blekhman_18.txt)」中のヒトサンプル名と同じにしています。
58,037 genes×6 samplesからなる出力ファイルはsrp001558_count_hoge6.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "srp001558_count_hoge6.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
uge <- assays(rse)$counts
dim(uge)
head(uge)
uge <- as.data.frame(uge)
data <- cbind(
uge$SRR032116,
uge$SRR032118 + uge$SRR032119,
uge$SRR032120 + uge$SRR032121,
uge$SRR032122 + uge$SRR032123,
uge$SRR032124 + uge$SRR032125,
uge$SRR032126 + uge$SRR032127)
colnames(data) <- c(
"HSF1", "HSF2", "HSF3",
"HSM1", "HSM2", "HSM3")
rownames(data)<- rownames(uge)
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題5と基本的に同じで、scale_counts関数実行前後の違いを表示させているだけです。
58,037 genes×11 samplesからなる出力ファイルはhoge7.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge7.txt"
library(recount)
load(in_f)
rse <- rse_gene
colSums(assays(rse)$counts)
rse <- scale_counts(rse)
colSums(assays(rse)$counts)
data <- assays(rse)$counts
colnames(data)<- colData(rse)$title
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題5の発展形として、recount(R package)
のrecount quick start guideのHTML
で書かれているgetRPKM関数実行結果を返すやり方です。58,037 genes×11 samplesからなる出力ファイルはhoge8.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge8.txt"
library(recount)
load(in_f)
rse <- rse_gene
colSums(assays(rse)$counts)
data <- getRPKM(rse)
colSums(data)
colnames(data)<- colData(rse)$title
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題8をベースとし、さらに例題4の発展形として、rowRanges関数実行結果に含まれるfeatureのより詳細なメタデータ情報
(遺伝子の染色体上の位置情報などを出力する)をファイルに保存しています。
出力ファイルはhoge9_counts.txtとhoge9_features.txtです。
in_f <- "rse_gene.Rdata"
out_f1 <- "hoge9_counts.txt"
out_f2 <- "hoge9_features.txt"
library(recount)
load(in_f)
rse <- rse_gene
colSums(assays(rse)$counts)
data <- getRPKM(rse)
colSums(data)
colnames(data)<- colData(rse)$title
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- rowRanges(rse)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP001540 | recount(Collado-Torres_2017)
recountパッケージを用いて、
SRP001540(Pickrell et al., Nature, 2010;ブラウザはIE以外を推奨)
のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、カウントデータの数値行列にした状態で保存するやり方を示します。
原著論文では、ヒトの69 lymphoblastoid cell linesのRNA-seqデータを取得しています。
このデータはAbsFilterGSEA
(Yoon et al., PLoS One, 2016)および
GSVA
(Hänzelmann et al., BMC Bioinformatics, 2013)中で、
検証用データとして用いられています。
具体的には、MSigDB C1で、
2つのsex-specificな遺伝子セット(chryq11とchrxp22)が発現変動しているという結果を得ているようです。
69 samplesの内訳は40 females and 29 malesのようですが、SRP001540
からは性別情報は不明です。原著論文(Pickrell et al., Nature, 2010)をよく読むと
http://eqtl.uchicago.eduからも情報取得可能だと書かれていますが性別情報までは探しきれませんでした。
Yale sequencing centerとArgonne sequencing centerの2か所でデータがとられており、
個体ごとに少なくとも2反復のデータがとられているようです(at least two replicate lanes per individual)。
性別(Gender)情報についてはその後、ウェブサイトrecount2上でSRP001540で検索し、
phenotype列のリンク先(SRP001540.tsv)から得られるcell lineのID情報を、
https://www.coriell.org/上で一つ一つ調べることで得られることがわかりました。
SRP001540.tsvの右側に
gender情報を追加したExcelファイルは、SRP001540_gender.xlsxです。
2018年8月7日に、recountWorkflow
で推奨されているscale_counts関数実行後のカウントデータとなるように変更しました。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
SRP001540という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でSRP001540で検索し、
gene列のRSE v2をダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "SRP001540"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
例題1で得られたファイル(rse_gene.Rdata; 約16MB)を読み込んで、
カウントの数値行列情報(58,037 genes×160 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge2.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題2と基本的に同じですが、列名を"cell line ID"と"sequencing center名"をマージした情報に置換しています。
出力ファイルはhoge3.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
colnames(data)
rownames(colData(rse))
identical(colnames(data), rownames(colData(rse)))
colnames(data) <- colData(rse)$title
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じですが、Yale sequence centerのデータのみ抽出し、technical replicatesをマージし、
40 Femalessと29 Malesが明瞭に分離されるようにソートしています。
in_f <- "rse_gene.Rdata"
out_f <- "hoge4.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
x <- assays(rse)$counts
x <- as.data.frame(x)
data <- cbind(x$SRR031822, x$SRR031953 + x$SRR031873,
x$SRR031952 + x$SRR031871, x$SRR031868,
x$SRR031819, x$SRR031897 + x$SRR031857,
x$SRR031823, x$SRR031959, x$SRR031955,
x$SRR031954, x$SRR031956, x$SRR031838,
x$SRR031918, x$SRR031817, x$SRR031949 + x$SRR031852,
x$SRR031841, x$SRR031865, x$SRR031896,
x$SRR031853, x$SRR031820, x$SRR031874,
x$SRR031895, x$SRR031870, x$SRR031839,
x$SRR031958, x$SRR031867, x$SRR031848,
x$SRR031847, x$SRR031818, x$SRR031919,
x$SRR031866, x$SRR031849, x$SRR031877,
x$SRR031814, x$SRR031914, x$SRR031812,
x$SRR031842, x$SRR031843, x$SRR031860, x$SRR031837,
x$SRR031917, x$SRR031821 + x$SRR031898,
x$SRR031950 + x$SRR031850, x$SRR031876 + x$SRR031862,
x$SRR031875, x$SRR031915, x$SRR031878 + x$SRR031863,
x$SRR031869, x$SRR031864, x$SRR031845,
x$SRR031951 + x$SRR031851, x$SRR031846,
x$SRR031916, x$SRR031844, x$SRR031813,
x$SRR031894, x$SRR031854, x$SRR031858,
x$SRR031859, x$SRR031872, x$SRR031816,
x$SRR031815, x$SRR031920 + x$SRR031899,
x$SRR031957 + x$SRR031855, x$SRR031840,
x$SRR031948, x$SRR031893, x$SRR031811, x$SRR031861)
colnames(data) <- c(paste("Female", 1:40, sep=""), paste("Male", 1:29, sep=""))
rownames(data) <- rownames(x)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じですが、Argonne sequence centerのデータのみ抽出し、
technical replicatesをマージし、40 Femalessと29 Malesが明瞭に分離されるようにソートしています。
in_f <- "rse_gene.Rdata"
out_f <- "hoge5.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
x <- assays(rse)$counts
x <- as.data.frame(x)
data <- cbind(x$SRR031834, x$SRR031962 + x$SRR031903,
x$SRR031964 + x$SRR031902, x$SRR031904,
x$SRR031879, x$SRR031926 + x$SRR031925,
x$SRR031880, x$SRR031941, x$SRR031944,
x$SRR031945, x$SRR031907, x$SRR031885,
x$SRR031946, x$SRR031831, x$SRR031940 + x$SRR031967,
x$SRR031889, x$SRR031931, x$SRR031927,
x$SRR031968, x$SRR031883, x$SRR031912,
x$SRR031934, x$SRR031901, x$SRR031882,
x$SRR031960, x$SRR031900, x$SRR031836,
x$SRR031832, x$SRR031833, x$SRR031937,
x$SRR031932, x$SRR031884, x$SRR031942,
x$SRR031828, x$SRR031936, x$SRR031835,
x$SRR031888, x$SRR031908, x$SRR031824, x$SRR031886,
x$SRR031938, x$SRR031881 + x$SRR031933,
x$SRR031965, x$SRR031909 + x$SRR031928,
x$SRR031911, x$SRR031947, x$SRR031913 + x$SRR031929,
x$SRR031906, x$SRR031930, x$SRR031891,
x$SRR031963 + x$SRR031966, x$SRR031890,
x$SRR031935, x$SRR031892, x$SRR031826,
x$SRR031921, x$SRR031969, x$SRR031922,
x$SRR031923, x$SRR031905, x$SRR031827,
x$SRR031830, x$SRR031943 + x$SRR031910,
x$SRR031939 + x$SRR031970, x$SRR031887,
x$SRR031961 + x$SRR031971, x$SRR031924,
x$SRR031829, x$SRR031825)
colnames(data) <- c(paste("Female", 1:40, sep=""), paste("Male", 1:29, sep=""))
rownames(data) <- rownames(x)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題4と基本的に同じですが、さらにサンプルのメタデータ情報ファイル(srp001558_meta_samples.txt)と、
遺伝子(features)のメタデータ情報ファイル(srp001540_meta_features.txt)も出力するやり方です。
他の出力ファイルはsrp001540_count_yale.txtです。
in_f <- "rse_gene.Rdata"
out_f1 <- "srp001540_count_yale.txt"
out_f2 <- "srp001540_meta_samples.txt"
out_f3 <- "srp001540_meta_features.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
x <- assays(rse)$counts
x <- as.data.frame(x)
data <- cbind(x$SRR031822, x$SRR031953 + x$SRR031873,
x$SRR031952 + x$SRR031871, x$SRR031868,
x$SRR031819, x$SRR031897 + x$SRR031857,
x$SRR031823, x$SRR031959, x$SRR031955,
x$SRR031954, x$SRR031956, x$SRR031838,
x$SRR031918, x$SRR031817, x$SRR031949 + x$SRR031852,
x$SRR031841, x$SRR031865, x$SRR031896,
x$SRR031853, x$SRR031820, x$SRR031874,
x$SRR031895, x$SRR031870, x$SRR031839,
x$SRR031958, x$SRR031867, x$SRR031848,
x$SRR031847, x$SRR031818, x$SRR031919,
x$SRR031866, x$SRR031849, x$SRR031877,
x$SRR031814, x$SRR031914, x$SRR031812,
x$SRR031842, x$SRR031843, x$SRR031860, x$SRR031837,
x$SRR031917, x$SRR031821 + x$SRR031898,
x$SRR031950 + x$SRR031850, x$SRR031876 + x$SRR031862,
x$SRR031875, x$SRR031915, x$SRR031878 + x$SRR031863,
x$SRR031869, x$SRR031864, x$SRR031845,
x$SRR031951 + x$SRR031851, x$SRR031846,
x$SRR031916, x$SRR031844, x$SRR031813,
x$SRR031894, x$SRR031854, x$SRR031858,
x$SRR031859, x$SRR031872, x$SRR031816,
x$SRR031815, x$SRR031920 + x$SRR031899,
x$SRR031957 + x$SRR031855, x$SRR031840,
x$SRR031948, x$SRR031893, x$SRR031811, x$SRR031861)
colnames(data) <- c(paste("Female", 1:40, sep=""), paste("Male", 1:29, sep=""))
rownames(data) <- rownames(x)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
#tmp <- colData(rse)
tmp <- apply(colData(rse), 2, as.character)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
#tmp <- rowData(rse)
tmp <- apply(rowData(rse), 2, as.character)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
例題5と基本的に同じですが、さらにサンプルのメタデータ情報ファイル(srp001558_meta_samples.txt)と、
遺伝子(features)のメタデータ情報ファイル(srp001540_meta_features.txt)も出力するやり方です。
他の出力ファイルはsrp001540_count_argonne.txtです。
in_f <- "rse_gene.Rdata"
out_f1 <- "srp001540_count_argonne.txt"
out_f2 <- "srp001540_meta_samples.txt"
out_f3 <- "srp001540_meta_features.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
x <- assays(rse)$counts
x <- as.data.frame(x)
data <- cbind(x$SRR031834, x$SRR031962 + x$SRR031903,
x$SRR031964 + x$SRR031902, x$SRR031904,
x$SRR031879, x$SRR031926 + x$SRR031925,
x$SRR031880, x$SRR031941, x$SRR031944,
x$SRR031945, x$SRR031907, x$SRR031885,
x$SRR031946, x$SRR031831, x$SRR031940 + x$SRR031967,
x$SRR031889, x$SRR031931, x$SRR031927,
x$SRR031968, x$SRR031883, x$SRR031912,
x$SRR031934, x$SRR031901, x$SRR031882,
x$SRR031960, x$SRR031900, x$SRR031836,
x$SRR031832, x$SRR031833, x$SRR031937,
x$SRR031932, x$SRR031884, x$SRR031942,
x$SRR031828, x$SRR031936, x$SRR031835,
x$SRR031888, x$SRR031908, x$SRR031824, x$SRR031886,
x$SRR031938, x$SRR031881 + x$SRR031933,
x$SRR031965, x$SRR031909 + x$SRR031928,
x$SRR031911, x$SRR031947, x$SRR031913 + x$SRR031929,
x$SRR031906, x$SRR031930, x$SRR031891,
x$SRR031963 + x$SRR031966, x$SRR031890,
x$SRR031935, x$SRR031892, x$SRR031826,
x$SRR031921, x$SRR031969, x$SRR031922,
x$SRR031923, x$SRR031905, x$SRR031827,
x$SRR031830, x$SRR031943 + x$SRR031910,
x$SRR031939 + x$SRR031970, x$SRR031887,
x$SRR031961 + x$SRR031971, x$SRR031924,
x$SRR031829, x$SRR031825)
colnames(data) <- c(paste("Female", 1:40, sep=""), paste("Male", 1:29, sep=""))
rownames(data) <- rownames(x)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
#tmp <- colData(rse)
tmp <- apply(colData(rse), 2, as.character)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
#tmp <- rowData(rse)
tmp <- apply(rowData(rse), 2, as.character)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | SRP001540 | GSVAdata(Hänzelmann_2013)
GSVAdataパッケージを用いて、
SRP001540(Pickrell et al., Nature, 2010;ブラウザはIE以外を推奨)
のカウント情報を含むExpressionSetオブジェクトという形式のデータセット(commonPickrellHuangという名前で格納されています)をdata関数を用いてロードしたり、カウントデータの数値行列にした状態で保存するやり方を示します。
原著論文では、ヒトの69個体(40 female samples and 29 male samples)のカウントデータを取得しています。
このデータはAbsFilterGSEA
(Yoon et al., PLoS One, 2016)、
およびGSVA
(Hänzelmann et al., BMC Bioinformatics, 2013)中で、
検証用データとして用いられています。具体的には、MSigDB C1で、
2つのsex-specificな遺伝子セット(chryq11とchrxp22)が発現変動しているという結果を得ているようです。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. pickrellCountsArgonneCQNcommon_esetの場合:
原著論文(Pickrell et al., Nature, 2010)中で記載されている、
Argonne sequencing centerで取得された正規化後のカウントデータのExpressionSetオブジェクトです。
library(GSVAdata)
data(commonPickrellHuang)
ls()
eset <- pickrellCountsArgonneCQNcommon_eset
eset
2. pickrellCountsYaleCQNcommon_esetの場合:
原著論文(Pickrell et al., Nature, 2010)中で記載されている、
Yale sequencing centerで取得された正規化後のカウントデータのExpressionSetオブジェクトです。計36サンプル。
library(GSVAdata)
data(commonPickrellHuang)
ls()
eset <- pickrellCountsYaleCQNcommon_eset
eset
3. pickrellCountsYaleCQNcommon_esetの場合:
例題2の続きで、11,508 genes (正確にはEntrez Gene IDs) × 36 samplesからなるカウントデータ行列をファイルに保存するやり方です。
このデータの最初のEntrez Gene IDは8567で、Gene symbolはMADDであることなどがわかります。
out_f <- "hoge3.txt"
library(GSVAdata)
data(commonPickrellHuang)
ls()
eset <- pickrellCountsYaleCQNcommon_eset
eset
write.exprs(eset, file=out_f)
4. pickrellCountsYaleCQNcommon_esetの場合:
例題3と基本的に同じで、write.exprs関数を使わないやり方です。
out_f <- "hoge4.txt"
library(GSVAdata)
data(commonPickrellHuang)
ls()
eset <- pickrellCountsYaleCQNcommon_eset
eset
data <- exprs(eset)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. pickrellCountsYaleCQNcommon_esetの場合:
例題4をベースとして、列名のところを性別(Female or Male)に変更しています。
out_f <- "hoge5.txt"
library(GSVAdata)
data(commonPickrellHuang)
ls()
eset <- pickrellCountsYaleCQNcommon_eset
dim(pData(eset))
head(pData(eset))
pData(eset)$Gender
table(pData(eset)$Gender)
data <- exprs(eset)
colnames(data) <- pData(eset)$Gender
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
6. pickrellCountsYaleCQNcommon_esetの場合:
例題5をベースとして、列名の性別(Female or Male)でソートしています。最初の23列がFemale、残りの13列がMaleになっていることがわかります。
11,508 Entrez gene IDs×36 samplesのカウントデータです。出力ファイルはhoge6.txtです。
out_f <- "hoge6.txt"
library(GSVAdata)
data(commonPickrellHuang)
ls()
eset <- pickrellCountsYaleCQNcommon_eset
dim(pData(eset))
head(pData(eset))
pData(eset)$Gender
table(pData(eset)$Gender)
data <- exprs(eset)
colnames(data) <- pData(eset)$Gender
head(data[1:2,])
data <- data[, order(colnames(data))]
head(data[1:2,])
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
7. pickrellCountsYaleCQNcommon_esetの場合:
例題6をベースとして、重複したEntrez gene IDのものが存在するので、ユニークにしています。
11,482 Entrez gene IDs×36 samplesのカウントデータです。出力ファイルはhoge7.txtです。
out_f <- "hoge7.txt"
library(GSVAdata)
library(genefilter)
data(commonPickrellHuang)
ls()
eset <- pickrellCountsYaleCQNcommon_eset
dim(pData(eset))
head(pData(eset))
pData(eset)$Gender
table(pData(eset)$Gender)
length(rownames(exprs(eset)))
length(unique(rownames(exprs(eset))))
hoge <- nsFilter(eset, var.filter=F,
remove.dupEntrez=T)
eset <- hoge$eset
length(rownames(exprs(eset)))
data <- exprs(eset)
colnames(data) <- pData(eset)$Gender
head(data[1:2,])
data <- data[, order(colnames(data))]
head(data[1:2,])
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | リアルデータ | ERP000546 | recount(Collado-Torres_2017)
recountパッケージを用いて、
ERP000546(原著論文なし;ブラウザはIE以外を推奨)のカウント情報を含むRangedSummarizedExperimentクラスオブジェクトという形式の.Rdataをダウンロードしたり、
カウントデータの数値行列にした状態で保存するやり方を示します。
RangedSummarizedExperimentというのがよくわからないとは思いますが、この中にEnsemblなどのgene IDだけでなく
gene symbolsや配列長情報なども含まれているので何かと便利なのです。
2018年8月7日に、recountWorkflow
で推奨されているscale_counts関数実行後のカウントデータとなるように変更しました。
「ファイル」−「ディレクトリの変更」でダウンロードしたいディレクトリに移動し以下をコピペ。
1. geneレベルカウントデータ情報を得たい場合:
ERP000546という名前のフォルダが作成されます。
中にあるrse-gene.Rdataをロードして読み込むとrse-geneというオブジェクト名で取り扱えます。
ウェブサイトrecount2上でERP000546で検索し、
gene列のRSE v2をダウンロードして得られるrse_gene.Rdataと同じです。
param_ID <- "ERP000546"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
2. geneレベルカウントデータ情報を得たい場合:
1.の発展形として、ダウンロードも行い、さらにカウントの数値行列情報(58,037 genes×47 samples)のみをタブ区切りテキストファイルで保存するやり方です。
出力ファイルはhoge2.txtです。
out_f <- "hoge2.txt"
param_ID <- "ERP000546"
library(recount)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
ウェブサイトrecount2上でERP000546で検索し、
gene列のRSE v2のところからダウンロードして得られた
geneレベルカウントデータ(rse_gene.Rdata; 約8MB)を読み込んで、
カウントの数値行列情報のみをタブ区切りテキストファイルで保存するやり方です。出力ファイルはhoge3.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3とベースとして、さらにサンプルのメタデータ情報ファイル(erp000546_meta_samples.txt)と、
遺伝子(features)のメタデータ情報ファイル(erp000546_meta_features.txt)も出力するやり方です。
カウントデータファイル(hoge4_counts.txt)は列名をERR...からERS...に変更しています。
in_f <- "rse_gene.Rdata"
out_f1 <- "hoge4_counts.txt"
out_f2 <- "erp000546_meta_samples.txt"
out_f3 <- "erp000546_meta_features.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
colnames(data) <- colData(rse)$sample
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- colData(rse)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
tmp <- rowData(rse)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
例題4で得られたサンプルのメタデータ情報ファイル(erp000546_meta_samples.txt)中の
ERR...からERS...の情報を手がかりにして、erp000546_meta_samples_added.txtの1番右の列で示すような
「これがheartで、これがkidneyで...」という対応関係をENAで1つ1つ調べたもので置き換えています。
出力ファイルはhoge5.txtです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge5.txt"
library(recount)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
uge <- assays(rse)$counts
dim(uge)
head(uge)
uge <- as.data.frame(uge)
data <- cbind(
uge$ERR030885 + uge$ERR030893,
uge$ERR030894 + uge$ERR030886,
uge$ERR030874 + uge$ERR030901,
uge$ERR030868 + uge$ERR030869 + uge$ERR030871 + uge$ERR030863 + uge$ERR030862 + uge$ERR030870,
uge$ERR030890 + uge$ERR030882,
uge$ERR030897 + uge$ERR030878,
uge$ERR030859 + uge$ERR030867 + uge$ERR030861 + uge$ERR030866 + uge$ERR030860,
uge$ERR030891,
uge$ERR030892 + uge$ERR030884,
uge$ERR030872 + uge$ERR030903,
uge$ERR030875 + uge$ERR030900,
uge$ERR030889 + uge$ERR030881,
uge$ERR030864 + uge$ERR030865 + uge$ERR030857 + uge$ERR030858 + uge$ERR030856,
uge$ERR030902 + uge$ERR030873,
uge$ERR030877 + uge$ERR030898,
uge$ERR030895 + uge$ERR030887,
uge$ERR030876 + uge$ERR030899,
uge$ERR030888 + uge$ERR030880,
uge$ERR030896 + uge$ERR030879)
colnames(data) <- c(
"kidney", "heart", "ovary",
"mixture1", "brain", "lymphnode",
"mixture2", "breast", "colon",
"thyroid", "white_blood_cells",
"adrenal", "mixture3", "testes",
"prostate", "liver",
"skeletal_muscle", "adipose", "lung")
rownames(data)<- rownames(uge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | シミュレーションデータ | RNA-seq | について
RNA-seq発現変動解析は、一般にゲノム配列(またはトランスクリプトーム配列)にリードをマップし、 遺伝子領域、エクソン(exon)領域など
特定の領域にマップされたリード数をカウントして得られた「カウントデータ(count data; count matrix)」を入力として用います。
しかし、使い慣れないパッケージだと正しい手順で実行できたかどうか不明なため、不安を覚えることもあると思います。
1つの解決策は「これが発現変動の上位に来ていないと絶対におかしい!」というような人工的なデータを作成し、それを入力として試すのです。
TCCは、2,3, 多群間, multi-factorにも対応しています。
compcodeRは2群間のみだがrandom effectにも対応し、任意の割合のoutlierを導入することができます。
実験系ユーザで動作確認用程度であれば、様々な実験デザインに対応しているTCCで極端なデータを作成して入力として用いるので十分だと思います(2015年4月6日現在)。
Rパッケージ:
- TCC(評価系はAUCのみ):Sun et al., BMC Bioinformatics, 2013
- compcodeR(AUC以外に多数存在):Soneson C., Bioinformatics, 2014
- SimSeq(詳細はまだ追い切れず):Benidt and Nettleton, Bioinformatics, 2015
- Polyester(様々な実験デザインに対応できてそう):Frazee et al., Bioinformatics, 2015
- LPEseq(データ生成のみ):Gim et al., PLoS One, 2016
- countsimQC(作成というより評価系):Soneson and Robinson, Bioinformatics, 2018
- seqgendiff:Gerard D, BMC Bioinformatics, 2020
カウント情報取得 | シミュレーションデータ | RNA-seq | Technical rep.(ポアソン分布)
ポアソン分布(Poisson distribution)に従うデータとは、任意の値(λ)を与えたときに、分散がλとなるような分布になるようなデータのことです。
以下では、1.ポアソン分布の感覚をつかみながら、2.Marioni et al., Genome Res., 2008の実際のデータを
経験分布(empirical distribution of read countsに相当)として与えてシミュレーションカウントデータの作成を行います。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1-1. ポアソン分布の感覚をつかむ(初級):
任意のλ(> 0)を与え、任意の数の乱数を発生させ、その分散がλに近い値になっているかどうか調べる。
param1 <- 8
param2 <- 100
out <- rpois(param2, lambda=param1)
out
var(out)
1-2. ポアソン分布の感覚をつかむ(中級):
λを1から100までにし、各λの値について発生させる乱数の数を増やし、λの値ごとの平均と分散を計算した結果をプロット
out <- NULL
for(i in 1:100){
x <- rpois(n=2000, lambda=i)
out <- rbind(out, c(mean(x), var(x)))
}
colnames(out) <- c("MEAN", "VARIANCE")
plot(out)
param1 <- c(5, 60, 200)
param2 <- 10000
out <- rpois(param2*length(param1), lambda=param1)
hist(out)
2. シミュレーションデータの作成本番(7サンプル分の行列データとして作成):
サンプルデータ2のSupplementaryTable2_changed.txtの「kidney 5サンプル vs. liver 5サンプル」のデータに相当しますが、このうちkidneyのデータを入力として与えることにします。
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge2.txt"
param1 <- 7
tmp <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
tmp <- rowSums(tmp[,1:5])
RPM <- tmp*1000000/sum(tmp)
LAMBDA <- RPM[RPM > 0]
out <- NULL
for(i in 1:param1){
out <- cbind(out, rpois(n=length(LAMBDA), lambda=LAMBDA))
}
obj <- rowSums(out) > 0
out2 <- out[obj,]
MEAN <- apply(out2, 1, mean)
VARIANCE <- apply(out2, 1, var)
plot(MEAN, VARIANCE, log="xy")
grid(col="gray", lty="dotted")
tmp <- cbind(out2, LAMBDA[obj], MEAN, VARIANCE)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3. シミュレーションデータの作成本番(遺伝子数(行数)を任意に与える場合):
サンプルデータ2のSupplementaryTable2_changed.txtの「kidney 5サンプル vs. liver 5サンプル」のデータに相当しますが、このうちkidneyのデータを入力として与えることにします。
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge3.txt"
param1 <- 7
param_Ngene <- 5000
tmp <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
tmp <- rowSums(tmp[,1:5])
LAMBDA <- tmp[tmp > 0]
LAMBDA <- sample(LAMBDA, param_Ngene, replace=TRUE)
LAMBDA <- LAMBDA*1000000/sum(LAMBDA)
out <- NULL
for(i in 1:param1){
out <- cbind(out, rpois(n=length(LAMBDA), lambda=LAMBDA))
}
obj <- apply(out, 1, var) > 0
out2 <- out[obj,]
MEAN <- apply(out2, 1, mean)
VARIANCE <- apply(out2, 1, var)
plot(MEAN, VARIANCE, log="xy")
grid(col="gray", lty="dotted")
tmp <- cbind(LAMBDA, out)
colnames(tmp) <- c("LAMBDA", paste("replicate",1:param1,sep=""))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
4. シミュレーションデータの作成本番(G1群3サンプル vs. G2群3サンプルのデータで、全遺伝子の10%がG1群で2倍高発現というデータにしたい場合):
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge4.txt"
param_G1 <- 3
param_G2 <- 3
param_Ngene <- 10000
param_PDEG <- 0.1
param_FC <- 2
tmp <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
tmp <- rowSums(tmp[,1:5])
LAMBDA <- tmp[tmp > 0]
LAMBDA <- sample(LAMBDA, param_Ngene, replace=TRUE)
LAMBDA <- LAMBDA*1000000/sum(LAMBDA)
DEG_degree <- rep(1, param_Ngene)
DEG_degree[1:(param_Ngene*param_PDEG)] <- param_FC
DEG_posi <- DEG_degree == param_FC
LAMBDA_A <- LAMBDA*DEG_degree
LAMBDA_B <- LAMBDA
outA <- NULL
for(i in 1:param_G1){
outA <- cbind(outA, rpois(n=length(LAMBDA_A), lambda=LAMBDA_A))
}
outB <- NULL
for(i in 1:param_G2){
outB <- cbind(outB, rpois(n=length(LAMBDA_B), lambda=LAMBDA_B))
}
tmp <- cbind(outA, outB)
colnames(tmp) <- c(paste("G1_rep",1:param_G1,sep=""),paste("G2_rep",1:param_G2,sep=""))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
data <- tmp
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
param_xrange <- c(0.5, 10000)
plot(x_axis, y_axis, pch=20, cex=0.1, xlim=log2(param_xrange), xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)")
points(x_axis[DEG_posi], y_axis[DEG_posi], col="magenta", pch=20, cex=0.1)
grid(col="gray", lty="dotted")
legend("topright", c("DEG", "non-DEG"), col=c("magenta", "black"), pch=19)
5. シミュレーションデータの作成本番(G1群3サンプル vs. G2群3サンプルのデータで、全遺伝子の10%がDEG。DEGのうちの80%がG1群で高発現、残りの20%がG2群で高発現というデータにしたい場合):
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_Ngene <- 10000
param_PDEG <- 0.1
param_FC <- 2
param_PG1 <- 0.8
tmp <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
tmp <- rowSums(tmp[,1:5])
LAMBDA <- tmp[tmp > 0]
LAMBDA <- sample(LAMBDA, param_Ngene, replace=TRUE)
LAMBDA <- LAMBDA*1000000/sum(LAMBDA)
DEG_degree_A <- rep(1, param_Ngene)
DEG_degree_A[1:(param_Ngene*param_PDEG*param_PG1)] <- param_FC
LAMBDA_A <- LAMBDA*DEG_degree_A
DEG_degree_B <- rep(1, param_Ngene)
DEG_degree_B[(param_Ngene*param_PDEG*param_PG1+1):param_Ngene*param_PDEG)] <- param_FC
LAMBDA_B <- LAMBDA*DEG_degree_B
DEG_posi <- (DEG_degree_A*DEG_degree_B) > 1
outA <- NULL
for(i in 1:param_G1){
outA <- cbind(outA, rpois(n=length(LAMBDA_A), lambda=LAMBDA_A))
}
outB <- NULL
for(i in 1:param_G2){
outB <- cbind(outB, rpois(n=length(LAMBDA_B), lambda=LAMBDA_B))
}
tmp <- cbind(outA, outB)
colnames(tmp) <- c(paste("G1_rep",1:param_G1,sep=""),paste("G2_rep",1:param_G2,sep=""))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
data <- tmp
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
param_xrange <- c(0.5, 10000)
plot(x_axis, y_axis, pch=20, cex=0.1, xlim=log2(param_xrange), xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)")
points(x_axis[DEG_posi], y_axis[DEG_posi], col="magenta", pch=20, cex=0.1)
grid(col="gray", lty="dotted")
legend("topright", c("DEG", "non-DEG"), col=c("magenta", "black"), pch=19)
6. シミュレーションデータの作成本番:
Robinson and Oshlack, 2010のFig. 2のデータとほとんど同じものを作りたい場合。違いはG1群およびG2群でuniqueに発現しているものを入れてないだけ
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge6.txt"
param_G1 <- 1
param_G2 <- 1
param_Ngene <- 20000
param_PDEG <- 0.1
param_FC <- 2
param_PG1 <- 0.8
tmp <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
tmp <- rowSums(tmp[,1:5])
LAMBDA <- tmp[tmp > 0]
LAMBDA <- sample(LAMBDA, param_Ngene, replace=TRUE)
LAMBDA <- LAMBDA*1000000/sum(LAMBDA)
DEG_degree_A <- rep(1, param_Ngene)
DEG_degree_A[1:(param_Ngene*param_PDEG*param_PG1)] <- param_FC
LAMBDA_A <- LAMBDA*DEG_degree_A
DEG_degree_B <- rep(1, param_Ngene)
DEG_degree_B[(param_Ngene*param_PDEG*param_PG1+1):(param_Ngene*param_PDEG)] <- param_FC
LAMBDA_B <- LAMBDA*DEG_degree_B
DEG_posi <- (DEG_degree_A*DEG_degree_B) > 1
outA <- NULL
for(i in 1:param_G1){
outA <- cbind(outA, rpois(n=length(LAMBDA_A), lambda=LAMBDA_A))
}
outB <- NULL
for(i in 1:param_G2){
outB <- cbind(outB, rpois(n=length(LAMBDA_B), lambda=LAMBDA_B))
}
tmp <- cbind(outA, outB)
colnames(tmp) <- c(paste("G1_rep",1:param_G1,sep=""),paste("G2_rep",1:param_G2,sep=""))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
data <- tmp
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
param_xrange <- c(0.5, 10000)
plot(x_axis, y_axis, pch=20, cex=0.1, xlim=log2(param_xrange), xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)")
points(x_axis[DEG_posi], y_axis[DEG_posi], col="magenta", pch=20, cex=0.1)
grid(col="gray", lty="dotted")
legend("topright", c("DEG", "non-DEG"), col=c("magenta", "black"), pch=19)
カウント情報取得 | シミュレーションデータ | RNA-seq | Biological rep. | 基礎
誰が最初か今のところ把握していませんがBiological replicatesのカウントデータが負の二項分布(negative binomial distribution; NB分布)
に従うというのがこの業界のコンセンサスです。つまり、ポアソン分布よりももっとばらつき(dispersion)が大きいということを言っています。
ここではまず、NB modelの一般式としてVARIANCE = MEAN + φ×(MEAN)^2において、MEAN=10 and φ=0.1
のNBモデルに従う乱数を発生させて発現変動解析との関連のイメージをつかみます。
このときのVARIANCEの期待値はVARIANCE = 10 + 0.1×(10)^2 = 20となります。このほかにも様々な例を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. MEAN=10 and φ=0.1の場合(基本形):
(MEAN-VARIANCE plotをファイルpngファイルに保存しています)
out_f <- "hoge1.png"
param_MEAN <- 10
param_dispersion <- 0.1
param_Ngene <- 1000
param_G1 <- 3
param_G2 <- 3
param_fig <- c(380, 420)
rnbinom(n=100, mu=param_MEAN, size=1/param_dispersion)
hoge <- rnbinom(n=10000, mu=param_MEAN, size=1/param_dispersion)
var(hoge)
hoge1 <-param_Ngene*(param_G1 + param_G2)
hoge2 <- rnbinom(n=hoge1, mu=param_MEAN, size=1/param_dispersion)
data <- matrix(hoge2, nrow=param_Ngene)
head(data)
dim(data)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
MEAN <- apply(data, 1, mean)
VARIANCE <- apply(data, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="black")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
DEG <- c(rep(0, param_G1), rep(param_MEAN*2, param_G2))
DEG
mean(DEG)
var(DEG)
points(mean(DEG), var(DEG), col="red", cex=1.0, pch=20)
legend("topright", c("DEG", "non-DEG"), col=c("red", "black"), pch=20)
dev.off()
2. G1群2サンプル vs. G2群2サンプルのデータで、全遺伝子の15%がDEG。DEGのうちの20%がG1群で4倍高発現、残りの80%がG2群で4倍高発現というデータにしたい場合:
out_f <- "hoge2.txt"
param_G1 <- 2
param_G2 <- 2
param_Ngene <- 20000
param_PDEG <- 0.15
param_FC <- 4
param_PG1 <- 0.2
library(NBPSeq)
data(arab)
data <- arab
data.cl <- c(rep(1, 3), rep(2, 3))
RPM <- sweep(data, 2, 1000000/colSums(data), "*")
RPM_A <- RPM[,data.cl == 1]
RPM_B <- RPM[,data.cl == 2]
RPM_A <- RPM_A[apply(RPM_A, 1, var) > 0,]
RPM_B <- RPM_B[apply(RPM_B, 1, var) > 0,]
MEAN <- c(apply(RPM_A, 1, mean), apply(RPM_B, 1, mean))
VARIANCE <- c(apply(RPM_A, 1, var), apply(RPM_B, 1, var))
DISPERSION <- (VARIANCE - MEAN)/(MEAN*MEAN)
mean_disp_tmp <- cbind(MEAN, DISPERSION)
mean_disp_tmp <- mean_disp_tmp[mean_disp_tmp[,2] > 0,]
hoge <- sample(1:nrow(mean_disp_tmp), param_Ngene, replace=TRUE)
mean_disp <- mean_disp_tmp[hoge,]
mu <- mean_disp[,1]
DEG_degree_A <- rep(1, param_Ngene)
DEG_degree_A[1:(param_Ngene*param_PDEG*param_PG1)] <- param_FC
mu_A <- mu*DEG_degree_A
DEG_degree_B <- rep(1, param_Ngene)
DEG_degree_B[(param_Ngene*param_PDEG*param_PG1+1):(param_Ngene*param_PDEG)] <- param_FC
mu_B <- mu*DEG_degree_B
DEG_posi_org <- (DEG_degree_A*DEG_degree_B) > 1
nonDEG_posi_org <- (DEG_degree_A*DEG_degree_B) == 1
outA <- NULL
for(i in 1:param_G1){
outA <- cbind(outA, rnbinom(n=length(mu_A), mu=mu_A, size=1/mean_disp[,2]))
}
outB <- NULL
for(i in 1:param_G2){
outB <- cbind(outB, rnbinom(n=length(mu_B), mu=mu_B, size=1/mean_disp[,2]))
}
out <- cbind(outA, outB)
obj <- rowSums(out) > 0
RAW <- out[obj,]
DEG_posi <- DEG_posi_org[obj]
nonDEG_posi <- nonDEG_posi_org[obj]
tmp <- cbind(RAW, DEG_posi)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
data <- RAW
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
#param_xrange <- c(0.5, 10000)
#plot(x_axis, y_axis, pch=20, cex=0.1, xlim=log2(param_xrange), xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)")
plot(x_axis, y_axis, pch=20, cex=0.1, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)")
points(x_axis[DEG_posi], y_axis[DEG_posi], col="magenta", pch=20, cex=0.1)
grid(col="gray", lty="dotted")
legend("topright", c("DEG", "non-DEG"), col=c("magenta", "black"), pch=19)
3. G1群1サンプル vs. G2群1サンプルのデータで、全遺伝子の15%がDEG。DEGのうちの20%がG1群で4倍高発現、残りの80%がG2群で4倍高発現というデータにしたい場合:
out_f <- "hoge3.txt"
param_G1 <- 1
param_G2 <- 1
param_Ngene <- 20000
param_PDEG <- 0.15
param_FC <- 4
param_PG1 <- 0.2
library(NBPSeq)
data(arab)
data <- arab
data.cl <- c(rep(1, 3), rep(2, 3))
RPM <- sweep(data, 2, 1000000/colSums(data), "*")
RPM_A <- RPM[,data.cl == 1]
RPM_B <- RPM[,data.cl == 2]
RPM_A <- RPM_A[apply(RPM_A,1,var) > 0,]
RPM_B <- RPM_B[apply(RPM_B,1,var) > 0,]
MEAN <- c(apply(RPM_A, 1, mean), apply(RPM_B, 1, mean))
VARIANCE <- c(apply(RPM_A, 1, var), apply(RPM_B, 1, var))
DISPERSION <- (VARIANCE - MEAN)/(MEAN*MEAN)
mean_disp_tmp <- cbind(MEAN, DISPERSION)
mean_disp_tmp <- mean_disp_tmp[mean_disp_tmp[,2] > 0,]
hoge <- sample(1:nrow(mean_disp_tmp), param_Ngene, replace=TRUE)
mean_disp <- mean_disp_tmp[hoge,]
mu <- mean_disp[,1]
DEG_degree_A <- rep(1, param_Ngene)
DEG_degree_A[1:(param_Ngene*param_PDEG*param_PG1)] <- param_FC
mu_A <- mu*DEG_degree_A
DEG_degree_B <- rep(1, param_Ngene)
DEG_degree_B[(param_Ngene*param_PDEG*param_PG1+1):(param_Ngene*param_PDEG)] <- param_FC
mu_B <- mu*DEG_degree_B
DEG_posi_org <- (DEG_degree_A*DEG_degree_B) > 1
nonDEG_posi_org <- (DEG_degree_A*DEG_degree_B) == 1
outA <- NULL
for(i in 1:param_G1){
outA <- cbind(outA, rnbinom(n=length(mu_A), mu=mu_A, size=1/mean_disp[,2]))
}
outB <- NULL
for(i in 1:param_G2){
outB <- cbind(outB, rnbinom(n=length(mu_B), mu=mu_B, size=1/mean_disp[,2]))
}
out <- cbind(outA, outB)
obj <- rowSums(out) > 0
RAW <- out[obj,]
DEG_posi <- DEG_posi_org[obj]
nonDEG_posi <- nonDEG_posi_org[obj]
tmp <- cbind(RAW, DEG_posi)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
data <- RAW
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
#param_xrange <- c(0.5, 10000)
#plot(x_axis, y_axis, pch=20, cex=0.1, xlim=log2(param_xrange), xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)")
plot(x_axis, y_axis, pch=20, cex=0.1, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)")
points(x_axis[DEG_posi], y_axis[DEG_posi], col="magenta", pch=20, cex=0.1)
grid(col="gray", lty="dotted")
legend("topright", c("DEG", "non-DEG"), col=c("magenta", "black"), pch=19)
- TCC:Sun et al., BMC Bioinformatics, 2013
- NBPSeq:Di et al., SAGMB, 2011
- GENE-counter:Cumbie et al., PLoS One, 2011
- DESeq:Anders and Huber, Genome Biol., 2010
- edgeR:Robinson et al., Bioinformatics, 2010
カウント情報取得 | シミュレーションデータ | RNA-seq | Biological rep. | 2群間 | 基礎 | LPEseq(Gim_2016)
LPEseqパッケージ中のgenerateData関数を用いて
2群間比較用のシミュレーションカウントデータを作成するやり方を示します。
群ごとの反復数は同じですので、例えば計6 samplesの場合はG1群3サンプル、G2群3サンプルになります。
従って、反復数はparam_Nrepで与えています。
発現変動の度合いに相当するパラメータは、(TCCではfold-changeで与えましたが)
LPEseqはカウント情報の差(count value difference)としてparam_effオプションで与えています。
その他、バラツキの度合いを表すdispersionパラメータは、param_dispオプションで与えています。
param_dispに0を与えるとPoisson分布と同じになり、大きな値を与えるほど群間のバラツキが大きくなります。
パッケージのインストールがまだの場合は、最初に例題0のやり方に従ってインストールをしておく必要があります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
0. パッケージのインストール:
最新版のファイル(2018年7月現在はLPEseq_v0.99.1.tar.gz)を
作業ディレクトリ上に予めダウンロードしておいてから、下記をコピペしてください。
source("http://statgen.snu.ac.kr/software/LPEseq/LPEseq.R")
install.packages("LPEseq_v0.99.1.tar.gz", repos=NULL, type="source")
1. 20,000 genes×6 samplesのシミュレーションデータを作成する場合:
Gim et al., 2016の
Table 2の、
Effect size = 1000, Dispersion = 0.25, # of DEGs = 2000の
シミュレーション条件に相当します。デフォルトではDEGの位置はランダムなので、param_NDEGで指定した最初の
2000行分がDEGになるようにソートさせています。
NB_parameter.txtという
経験分布(empirical distribution)情報ファイルを内部的に読み込んでいます。
out_f <- "hoge1.txt"
param_Nrep <- 3
param_Ngene <- 20000
param_eff <- 1000
param_disp <- 0.25
param_NDEG <- 2000
library(LPEseq)
set.seed(1000)
hoge <- generateData(n.rep=param_Nrep,
n.gene=param_Ngene,
n.deg=param_NDEG,
eff=param_eff,
disp=param_disp)
hoge <- hoge[order(hoge[,ncol(hoge)], decreasing=T), ]
DEG_posi <- hoge[, ncol(hoge)]
data <- hoge[, -ncol(hoge)]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. 20,000 genes×2 samplesのシミュレーションデータを作成する場合:
例題1と基本的に同じで、param_Nrepで指定する反復数の部分のみが異なります。
シミュレーション条件がGim et al., 2016の
Table 1の、
Effect size = 1000, Dispersion = 0.25, # of DEGs = 2000に相当します。
out_f <- "hoge2.txt"
param_Nrep <- 1
param_Ngene <- 20000
param_eff <- 1000
param_disp <- 0.25
param_NDEG <- 2000
library(LPEseq)
set.seed(1000)
hoge <- generateData(n.rep=param_Nrep,
n.gene=param_Ngene,
n.deg=param_NDEG,
eff=param_eff,
disp=param_disp)
hoge <- hoge[order(hoge[,ncol(hoge)], decreasing=T), ]
DEG_posi <- hoge[, ncol(hoge)]
data <- hoge[, -ncol(hoge)]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3. 20,000 genes×2 samplesのシミュレーションデータを作成する場合:
例題2と同じ反復なしデータですが、シミュレーション条件が
Gim et al., 2016の
Table 1の、
Effect size = 500, Dispersion = 0.01, # of DEGs = 1000に相当します。
out_f <- "hoge3.txt"
param_Nrep <- 1
param_Ngene <- 20000
param_eff <- 500
param_disp <- 0.01
param_NDEG <- 1000
library(LPEseq)
set.seed(1000)
hoge <- generateData(n.rep=param_Nrep,
n.gene=param_Ngene,
n.deg=param_NDEG,
eff=param_eff,
disp=param_disp)
hoge <- hoge[order(hoge[,ncol(hoge)], decreasing=T), ]
DEG_posi <- hoge[, ncol(hoge)]
data <- hoge[, -ncol(hoge)]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
4. 20,000 genes×6 samplesのシミュレーションデータを作成する場合:
例題3と同じシミュレーション条件で、例題1と同じ反復ありデータです。
out_f <- "hoge4.txt"
param_Nrep <- 3
param_Ngene <- 20000
param_eff <- 500
param_disp <- 0.01
param_NDEG <- 1000
library(LPEseq)
set.seed(1000)
hoge <- generateData(n.rep=param_Nrep,
n.gene=param_Ngene,
n.deg=param_NDEG,
eff=param_eff,
disp=param_disp)
hoge <- hoge[order(hoge[,ncol(hoge)], decreasing=T), ]
DEG_posi <- hoge[, ncol(hoge)]
data <- hoge[, -ncol(hoge)]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | シミュレーションデータ | RNA-seq | Biological rep. | 2群間 | 基礎 | TCC(Sun_2013)
TCCパッケージ中のsimulateReadCounts関数を用いて
2群間比較用のシミュレーションカウントデータを作成するやり方を示します。
特に、以下に示すようなTbT論文(Kadota et al., Algorithms Mol. Biol., 2012)
の図1とほぼ同じシミュレーション条件に固定して、TCCパッケージの使い方に慣れることに重点を置きます:
G1群 vs. G2群で各群の反復数を3に固定、 、つまり「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3」の計6サンプル。
全10,000遺伝子、最初の20%分(gene_1〜gene_2000)が発現変動遺伝子(DEG)、
全2,000 DEGsの内訳:最初の90%分(gene_1〜gene_1800)がG1群で4倍高発現、残りの10%分(gene_1801〜gene_2000)がG2群で4倍高発現。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータを作成する場合:
基本形。
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_Ngene <- 10000
param_PDEG <- 0.2
param_FC <- 4
param_PG1 <- 0.9
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=c(param_PG1, 1-param_PG1),
DEG.foldchange=c(param_FC, param_FC),
replicates=c(param_G1, param_G2))
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータを作成する場合:
1. と基本的に同じですが、set.seed(1000)の行頭に#を入れてコメントアウトしているので、得られる数値自体は毎回変わるようになります。
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_Ngene <- 10000
param_PDEG <- 0.2
param_FC <- 4
param_PG1 <- 0.9
library(TCC)
#set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=c(param_PG1, 1-param_PG1),
DEG.foldchange=c(param_FC, param_FC),
replicates=c(param_G1, param_G2))
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータを作成する場合:
1.と同じですが、記述形式を変えてます。
out_f <- "hoge3.txt"
param_replicates <- c(3, 3)
param_Ngene <- 10000
param_PDEG <- 0.2
param_FC <- c(4, 4)
param_DEGassign <- c(0.9, 0.1)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
4. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータを作成する場合:
2. と基本的に同じですが、set.seed(1000)の行頭に#を入れてコメントアウトしているので、得られる数値自体は毎回変わるようになります。
out_f <- "hoge4.txt"
param_replicates <- c(3, 3)
param_Ngene <- 10000
param_PDEG <- 0.2
param_FC <- c(4, 4)
param_DEGassign <- c(0.9, 0.1)
library(TCC)
#set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータを作成する場合:
3.と同じですが、それにプラスアルファの情報として、シミュレーション条件の概要が分かるようにpseudo-colorイメージのpngファイルを生成しています。
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5.png"
param_replicates <- c(3, 3)
param_Ngene <- 10000
param_PDEG <- 0.2
param_FC <- c(4, 4)
param_DEGassign <- c(0.9, 0.1)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
6. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータを作成する場合:
3.と同じですが、それにプラスアルファの情報として、M-A plotのpngファイルを生成しています。
out_f1 <- "hoge6.txt"
out_f2 <- "hoge6.png"
param_replicates <- c(3, 3)
param_Ngene <- 10000
param_PDEG <- 0.2
param_FC <- c(4, 4)
param_DEGassign <- c(0.9, 0.1)
param_fig <- c(602, 450)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, normalize=F, median.lines=T, col=c("black","red"))
dev.off()
7. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータを作成する場合:
6.と同じですが、それにプラスアルファの情報として、シミュレーション条件の概要を一望できるpseudo-colorイメージのpngファイルも生成しています。
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7_MAplot.png"
out_f3 <- "hoge7_pseudo.png"
param_replicates <- c(3, 3)
param_Ngene <- 10000
param_PDEG <- 0.2
param_FC <- c(4, 4)
param_DEGassign <- c(0.9, 0.1)
param_fig2 <- c(602, 450)
param_fig3 <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig2[1], height=param_fig2[2])
plot(tcc, normalize=F, median.lines=T, col=c("black","red"))
dev.off()
png(out_f3, pointsize=13, width=param_fig3[1], height=param_fig3[2])
plotFCPseudocolor(tcc)
dev.off()
カウント情報取得 | シミュレーションデータ | RNA-seq | Biological rep. | 2群間 | 応用 | TCC(Sun_2013)
TCCパッケージ中のsimulateReadCounts関数を用いて
2群間比較用のシミュレーションカウントデータを作成するやり方を示します。
ここでは、基礎の7.のコードを基本として、各群の反復数、全遺伝子数、発現変動遺伝子(DEG)数の割合、各群のDEGの発現変動の度合い、DEG数の各群への割り振りをいろいろ変えます。
試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(1000)の前に#を入れましょう。つまり「set.seed(1000)」->「#set.seed(1000)」です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 12,500 genes×6 samplesのシミュレーションデータを作成する場合:
全遺伝子数(12500)、反復数(G1が2, G2が4)、DEGの割合が0.32 (12500*0.32=4000)、
DEGの発現変動の度合い(G1が5.3, G2が0.25)、DEG数の各群への割り振り(G1が0.2, G2が0.8)。
これは全部で4,000 DEGsのうち、gene_1からgene_800までがG1群で5.3倍高発現、gene_801からgene_4000までがG2群で0.25倍高発現(つまりG1群で4倍高発現)を意味し、
事実上全DEGがG1群で高発現というシミュレーション条件になります。M-A plot実行時にmedian.lines=Fとして、水平線や右側に数値が出ないようにしています。
out_f1 <- "hoge1.txt"
out_f2 <- "hoge1_MAplot.png"
out_f3 <- "hoge1_pseudo.png"
param_replicates <- c(2, 4)
param_Ngene <- 12500
param_PDEG <- 0.32
param_FC <- c(5.3, 0.25)
param_DEGassign <- c(0.2, 0.8)
param_fig2 <- c(602, 450)
param_fig3 <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig2[1], height=param_fig2[2])
plot(tcc, normalize=F, median.lines=F, col=c("black","red"))
dev.off()
png(out_f3, pointsize=13, width=param_fig3[1], height=param_fig3[2])
plotFCPseudocolor(tcc)
dev.off()
2. 20,023 genes×11 samplesのシミュレーションデータを作成する場合:
全遺伝子数(20023)、反復数(G1が4, G2が7)、DEGの割合が0.27、
DEGの発現変動の度合い(G1が3.2, G2が4.7)、DEG数の各群への割り振り(G1が0.65, G2が0.35)。
これは全部で5,406 DEGsのうち、gene_1からgene_3514までがG1群で3.2倍高発現、gene_3515からgene_5406までがG2群で4.7倍高発現を意味します。
M-A plot実行時にmedian.lines=Tとして、水平線や右側に数値が出るようにしています。
out_f1 <- "hoge2.txt"
out_f2 <- "hoge2_MAplot.png"
out_f3 <- "hoge2_pseudo.png"
param_replicates <- c(4, 7)
param_Ngene <- 20023
param_PDEG <- 0.27
param_FC <- c(3.2, 4.7)
param_DEGassign <- c(0.65, 0.35)
param_fig2 <- c(602, 450)
param_fig3 <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig2[1], height=param_fig2[2])
plot(tcc, normalize=F, median.lines=T, col=c("black","red"))
dev.off()
png(out_f3, pointsize=13, width=param_fig3[1], height=param_fig3[2])
plotFCPseudocolor(tcc)
dev.off()
3. 20,023 genes×2 samplesのシミュレーションデータを作成する場合:
全遺伝子数(20023)、反復数(G1が1, G2が1)、DEGの割合が0.27、
DEGの発現変動の度合い(G1が3.2, G2が4.7)、DEG数の各群への割り振り(G1が0.65, G2が0.35)。
これは全部で5,406 DEGsのうち、gene_1からgene_3514までがG1群で3.2倍高発現、gene_3515からgene_5406までがG2群で4.7倍高発現を意味します。
M-A plot実行時にmedian.lines=Tとして、水平線や右側に数値が出るようにしています。
out_f1 <- "hoge3.txt"
out_f2 <- "hoge3_MAplot.png"
out_f3 <- "hoge3_pseudo.png"
param_replicates <- c(1, 1)
param_Ngene <- 20023
param_PDEG <- 0.27
param_FC <- c(3.2, 4.7)
param_DEGassign <- c(0.65, 0.35)
param_fig2 <- c(602, 450)
param_fig3 <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig2[1], height=param_fig2[2])
plot(tcc, normalize=F, median.lines=T, col=c("black","red"))
dev.off()
png(out_f3, pointsize=13, width=param_fig3[1], height=param_fig3[2])
plotFCPseudocolor(tcc)
dev.off()
カウント情報取得 | シミュレーションデータ | RNA-seq | Biological rep. | 3群間 | 基礎 | TCC(Sun_2013)
TCCパッケージ中のsimulateReadCounts関数を用いて
3群間比較用のシミュレーションカウントデータを作成するやり方を示します。
ここでは、G1, G2, G3群で各群の反復数を3に固定、 、つまり「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3, G3_rep1, G3_rep2, G3_rep3」の計9サンプル。
全10,000遺伝子、最初の25%分(gene_1〜gene_2500)が発現変動遺伝子(DEG)。
試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(1000)の前に#を入れましょう。つまり「set.seed(1000)」->「#set.seed(1000)」です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 10,000 genes×9 samplesのシミュレーションデータを作成する場合:
全遺伝子数が10000、反復数(G1が3, G2が3, G3が3)、DEGの割合が0.25 (10000*0.25=2500)、
DEGの発現変動の度合い(G1が4, G2が4, G3が4)、DEG数の各群への割り振り(G1が1/3, G2が1/3, G3が1/3)。
これは全部で2,500 DEGsのうち、gene_1からgene_833までがG1群で4倍高発現、gene_834からgene_1666までがG2群で4倍高発現、gene_1667からgene_2500までがG3群で4倍高発現を意味します。
out_f1 <- "hoge1.txt"
out_f2 <- "hoge1.png"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.25
param_FC <- c(4, 4, 4)
param_DEGassign <- c(1/3, 1/3, 1/3)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
2. 10,000 genes×9 samplesのシミュレーションデータを作成する場合:
全遺伝子数が10000、反復数(G1が3, G2が3, G3が3)、
DEGの割合が0.25 (10000*0.25=2500)、
DEGの発現変動の度合い(G1が4, G2が4, G3が4)、
DEG数の各群への割り振り(G1が0.5, G2が0.3, G3が0.2)。
これは全部で2,500 DEGsのうち、gene_1からgene_1250までがG1群で4倍高発現、gene_1251からgene_2000までがG2群で4倍高発現、gene_2001からgene_2500までがG3群で4倍高発現を意味します。
out_f1 <- "hoge2.txt"
out_f2 <- "hoge2.png"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.25
param_FC <- c(4, 4, 4)
param_DEGassign <- c(0.5, 0.3, 0.2)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
3. 10,000 genes×9 samplesのシミュレーションデータを作成する場合:
全遺伝子数が10000、反復数(G1が3, G2が3, G3が3)、
DEGの割合が0.25 (10000*0.25=2500)、
DEGの発現変動の度合い(G1が4, G2が4, G3が4)、
DEG数の各群への割り振り(G1が0.5, G2が0.4, G3が0.1)。
これは全部で2,500 DEGsのうち、gene_1からgene_1250までがG1群で4倍高発現、gene_1251からgene_2250までがG2群で4倍高発現、gene_2251からgene_2500までがG3群で4倍高発現を意味します。
out_f1 <- "hoge3.txt"
out_f2 <- "hoge3.png"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.25
param_FC <- c(4, 4, 4)
param_DEGassign <- c(0.5, 0.4, 0.1)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
4. 10,000 genes×9 samplesのシミュレーションデータを作成する場合:
全遺伝子数が10000、反復数(G1が3, G2が3, G3が3)、
DEGの割合が0.25 (10000*0.25=2500)、
DEGの発現変動の度合い(G1が4, G2が4, G3が4)、
DEG数の各群への割り振り(G1が0.6, G2が0.2, G3が0.2)。
これは全部で2,500 DEGsのうち、gene_1からgene_1500までがG1群で4倍高発現、gene_1501からgene_2000までがG2群で4倍高発現、gene_2001からgene_2500までがG3群で4倍高発現を意味します。
out_f1 <- "hoge4.txt"
out_f2 <- "hoge4.png"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.25
param_FC <- c(4, 4, 4)
param_DEGassign <- c(0.6, 0.2, 0.2)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
5. 10,000 genes×9 samplesのシミュレーションデータを作成する場合:
全遺伝子数が10000、反復数(G1が3, G2が3, G3が3)、
DEGの割合が0.25 (10000*0.25=2500)、
DEGの発現変動の度合い(G1が4, G2が4, G3が4)、
DEG数の各群への割り振り(G1が0.6, G2が0.3, G3が0.1)。
これは全部で2,500 DEGsのうち、gene_1からgene_1500までがG1群で4倍高発現、gene_1501からgene_2250までがG2群で4倍高発現、gene_2251からgene_2500までがG3群で4倍高発現を意味します。
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5.png"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.25
param_FC <- c(4, 4, 4)
param_DEGassign <- c(0.6, 0.3, 0.1)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
6. 10,000 genes×9 samplesのシミュレーションデータを作成する場合:
全遺伝子数が10000、反復数(G1が3, G2が3, G3が3)、
DEGの割合が0.25 (10000*0.25=2500)、
DEGの発現変動の度合い(G1が4, G2が4, G3が4)、
DEG数の各群への割り振り(G1が0.7, G2が0.2, G3が0.1)。
これは全部で2,500 DEGsのうち、gene_1からgene_1750までがG1群で4倍高発現、gene_1751からgene_2250までがG2群で4倍高発現、gene_2251からgene_2500までがG3群で4倍高発現を意味します。
out_f1 <- "hoge6.txt"
out_f2 <- "hoge6.png"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.25
param_FC <- c(4, 4, 4)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
7. 10,000 genes×9 samplesのシミュレーションデータを作成する場合:
全遺伝子数が10000、反復数(G1が3, G2が3, G3が3)、
DEGの割合が0.25 (10000*0.25=2500)、
DEGの発現変動の度合い(G1が4, G2が4, G3が4)、
DEG数の各群への割り振り(G1が0.8, G2が0.1, G3が0.1)。
これは全部で2,500 DEGsのうち、gene_1からgene_2000までがG1群で4倍高発現、gene_2001からgene_2250までがG2群で4倍高発現、gene_2251からgene_2500までがG3群で4倍高発現を意味します。
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7.png"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.25
param_FC <- c(4, 4, 4)
param_DEGassign <- c(0.8, 0.1, 0.1)
param_fig <- c(450, 600)
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotFCPseudocolor(tcc)
dev.off()
カウント情報取得 | シミュレーションデータ | scRNA-seq | について
single-cell RNA-seq (scRNA-seq)用のシミュレーションデータを作成するものです。
Rパッケージ:
- BASiCS:Vallejos et al., PLoS Comput Biol., 2015
- scDD:Korthauer et al., Genome Biol., 2016
- Splatter:Zappia et al., Genome Biol., 2017
- powsimR:Vieth et al., Bioinformatics, 2017
- countsimQC(作成というより評価系):Soneson and Robinson, Bioinformatics, 2018
- SymSim:Zhang et al., Nat Commun., 2019
カウント情報取得 | シミュレーションデータ | scRNA-seq | 基礎(同一細胞群) | Splatter(Zappia_2017)
Splatterパッケージ中を用いて
シミュレーションカウントデータを作成するやり方を示します。原著論文(Zappia et al., Genome Biol., 2017)
中に書かれていますが、Splatterは全部で6つのシミュレーションモデル(Simple, Lun, Lun 2, scDD, BASiCS, and Splat)を実装しています。
この項目ではSplatter原著論文中で新たに提案されたSplatというモデルに基づくシミュレーションデータを生成するやり方を示します。
様々な記述法があります。「基礎(同一細胞群)」の項目では、同一タイプの細胞(cells of same type; one cell type)からなるシミュレーションデータを生成しています。
従来型のRNA-seqカウントデータ解析を知っているヒト向けの説明としては、
「反復あり二群間比較用シミュレーションデータの中で、1つの群のみのデータを生成しており、反復数が100のようなものだ」
と解釈すればよいです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
splatSimulate関数のデフォルトのやり方です。
out_f <- "hoge1.txt"
library(splatter)
hoge <- splatSimulate()
hoge
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
例題1と同じ結果が得られます。splatSimulate関数実行時にmethod="single"をつけていますが、
singleがデフォルトであることがわかります。
out_f <- "hoge2.txt"
library(splatter)
hoge <- splatSimulate(method="single")
hoge
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
例題1-2と同じ結果が得られます。splatSimulateSingle関数をデフォルトで利用するやり方です。
つまり、splatSimulate(method="single")は、splatSimulateSingleと同じ意味だということです。
out_f <- "hoge3.txt"
library(splatter)
hoge <- splatSimulateSingle()
hoge
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
4. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
例題1-3と同じ結果が得られます。newSplatParams関数を用いてシミュレーション用パラメータを作成した結果を、
splatSimulateSingle関数実行時のオプションとして与えています。
out_f <- "hoge4.txt"
library(splatter)
params <- newSplatParams()
params
hoge <- splatSimulateSingle(params=params)
hoge
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
例題1-4と同じ結果が得られます。例題4と似ていますが、splatSimulateSingle関数の代わりにsplatSimulate関数を利用しています。
この場合、method="single"オプションをつけないといけませんが、それを例題2とは少し異なる方法で記載しているだけです。
out_f <- "hoge5.txt"
param_method <- "single"
library(splatter)
params <- newSplatParams()
params
hoge <- splatSimulate(
method=param_method,
params=params)
hoge
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
6. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
例題1-5と同じ結果が得られます。遺伝子数と細胞数を指定するオプションを明示するやり方です。
細胞数は、newSplatParams関数実行時にbatchCellsオプションのところで与えています
(newSplatParams関数実行時にnCellsオプションのところで与えるとエラーが出ます。)
out_f <- "hoge6.txt"
param_method <- "single"
param_Ngene <- 10000
param_Ncell <- 100
library(splatter)
params <- newSplatParams(
nGenes=param_Ngene,
batchCells=param_Ncell)
params
hoge <- splatSimulate(
method=param_method,
params=params)
hoge
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
7. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
例題1-6と同じ結果が得られます。例題6と似ていますが、newSplatParams関数を利用せずに、
直接splatSimulate関数内で遺伝子数と細胞数を指定するやり方です。
out_f <- "hoge7.txt"
param_method <- "single"
param_Ngene <- 10000
param_Ncell <- 100
library(splatter)
hoge <- splatSimulate(
method=param_method,
nGenes=param_Ngene,
batchCells=param_Ncell)
hoge
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
8. 10,000 genes×100 cellsのシミュレーションデータを作成する場合:
例題7と同じです。シミュレーションに用いたパラメータ情報を表示させるところを追加しただけです。
遺伝子についてはrowData(hoge)で取り出せます。splatter.html中のGene informationというのに相当します。
細胞についてはcolData(hoge)で取り出せます。Cell informationというのに相当します。
さらにassays(hoge)でその他の情報も取り出せます。これはGene by cell informationというのに相当しますが、
情報量が多すぎるのでstr(assays(hoge))を示してassays(hoge)の構造(structure)を表示させるに留めています。
out_f <- "hoge8.txt"
param_method <- "single"
param_Ngene <- 10000
param_Ncell <- 100
library(splatter)
hoge <- splatSimulate(
method=param_method,
nGenes=param_Ngene,
batchCells=param_Ncell)
hoge
rowData(hoge)
colData(hoge)
str(assays(hoge))
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | シミュレーションデータ | scRNA-seq | 基礎(異なる細胞群) | Splatter(Zappia_2017)
Splatterパッケージ中のSplatというモデルを用いて
シミュレーションカウントデータを作成するやり方を示します。「異なる細胞群」というのは、細胞の種類数(群数)が2つ以上あることを意味します。
従って、param_methodのところは事実上"groups"で固定です。
従来型のRNA-seqカウントデータ解析を知っているヒト向けの説明としては、例えば例題1は
「反復あり3群間比較用シミュレーションデータの中で、各群の反復数が概ね40のようなものだ」
と解釈すればよいです。概ねを強調している理由は、c(40, 40, 40)
という数値ベクトルを与えてはいるものの、実際にはその周辺の数(例えばGroup1が32個、Group2が38個、Group3が50個)が得られるからです。総数は不変のようです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
細胞の種類数(群数)は3つで、各種類(各群)につき40細胞という条件です。
細胞名がCell1, Cell2, ..., Cell120となってはいますが、colData(hoge)のGroup列の並びからも想像できるように、細胞の種類(群)ごとに並んでいるわけではないのでご注意ください。
table(colData(hoge)$Group)実行結果からもわかりますが、得られたデータは「Group1の細胞数が32個、Group2の細胞数が38個、Group3の細胞数が50個」みたいな感じになっていることがわかります。
out_f <- "hoge1.txt"
param_method <- "groups"
param_Ngene <- 10000
param_Ncell <- c(40, 40, 40)
library(splatter)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulate(
method=param_method,
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
data <- counts(hoge)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
例題1と基本的に同じですが、列名を(Group1, Group2, Group3などに)変更してから、細胞の種類(群)ごとにソートしています。
(遺伝子名の列を除く)最初の32列分がGroup1、次の38列分がGroup2、残りの50列分がGroup3みたいになっていることがわかります。
out_f <- "hoge2.txt"
param_method <- "groups"
param_Ngene <- 10000
param_Ncell <- c(40, 40, 40)
library(splatter)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulate(
method=param_method,
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
data <- counts(hoge)
colnames(data) <- colData(hoge)$Group
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
トータルの細胞数(=120)は同じですが、細胞の種類数(群数)は6つで、各種類(各群)につき20細胞という条件です。
「Group1が19列、Group2が16列、Group3が22列、Group4が19列、Group5が31列、そしてGroup6が13列」みたいになっていることがわかります。
out_f <- "hoge3.txt"
param_method <- "groups"
param_Ngene <- 10000
param_Ncell <- c(20, 20, 20, 20, 20, 20)
library(splatter)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulate(
method=param_method,
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
data <- counts(hoge)
colnames(data) <- colData(hoge)$Group
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
4. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
例題3と基本的に同じですが、gsub関数を用いて文字列"Group"を"G"に置換して、群名を「Group1, Group2, ...」から
「G1, G2, ...」に変更しています。
「G1が19列、G2が16列、G3が22列、G4が19列、G5が31列、そしてG6が13列」みたいになっていることがわかります。
out_f <- "hoge4.txt"
param_method <- "groups"
param_Ngene <- 10000
param_Ncell <- c(20, 20, 20, 20, 20, 20)
library(splatter)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulate(
method=param_method,
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
data <- counts(hoge)
colnames(data) <- colData(hoge)$Group
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
colnames(data) <- gsub("Group", "G", colnames(data))
colnames(data)
table(colnames(data))
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
例題4と基本的に同じですが、splatSimulate関数の代わりにsplatSimulateGroups関数を利用しています。
「G1が19列、G2が16列、G3が22列、G4が19列、G5が31列、そしてG6が13列」みたいになっていることがわかります。
out_f <- "hoge5.txt"
param_Ngene <- 10000
param_Ncell <- c(20, 20, 20, 20, 20, 20)
library(splatter)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulateGroups(
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
data <- counts(hoge)
colnames(data) <- colData(hoge)$Group
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
colnames(data) <- gsub("Group", "G", colnames(data))
colnames(data)
table(colnames(data))
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
カウント情報取得 | シミュレーションデータ | scRNA-seq | 応用(異なる細胞群) | Splatter(Zappia_2017)
Splatterパッケージ中のSplatというモデルを用いて
シミュレーションカウントデータを作成するやり方を示します。「応用(異なる細胞群)」では、正規化後のデータを取得したり、クラスタリングを直接行ったりする例を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
トータルの細胞数(=120)は同じですが、細胞の種類数(群数)は6つで、各種類(各群)につき20細胞という条件です。
G1が19列、G2が16列、G3が22列、G4が19列、G5が31列、そしてG6が13列となっていることがわかります。
「基礎(異なる細胞群)」の例題5をベースとして、
scaterパッケージ中のnormalize関数を実行した結果を出力しています。
log2変換後のデータです。
out_f <- "hoge1.txt"
param_Ngene <- 10000
param_Ncell <- c(20, 20, 20, 20, 20, 20)
library(splatter)
library(scater)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulateGroups(
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
sizeFactors(hoge) <- librarySizeFactors(hoge)
hoge <- normalize(hoge)
data <- logcounts(hoge)
colnames(data) <- colData(hoge)$Group
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
colnames(data) <- gsub("Group", "G", colnames(data))
colnames(data)
table(colnames(data))
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
例題1と基本的に同じですが、主成分分析(PCA)も行っています。
out_f <- "hoge2.txt"
param_Ngene <- 10000
param_Ncell <- c(20, 20, 20, 20, 20, 20)
library(splatter)
library(scater)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulateGroups(
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
sizeFactors(hoge) <- librarySizeFactors(hoge)
hoge <- normalize(hoge)
plotPCA(hoge, colour_by = "Group")
data <- logcounts(hoge)
colnames(data) <- colData(hoge)$Group
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
colnames(data) <- gsub("Group", "G", colnames(data))
colnames(data)
table(colnames(data))
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3. 10,000 genes×120 cellsのシミュレーションデータを作成する場合:
例題1と基本的に同じですが、t-distributed stochastic neighbour embedding (t-SNE)も行っています。
out_f <- "hoge3.txt"
param_Ngene <- 10000
param_Ncell <- c(20, 20, 20, 20, 20, 20)
library(splatter)
library(scater)
param_tmp <- param_Ncell/sum(param_Ncell)
hoge <- splatSimulateGroups(
nGenes=param_Ngene,
batchCells=sum(param_Ncell),
group.prob=param_tmp)
hoge
colData(hoge)
table(colData(hoge)$Group)
sizeFactors(hoge) <- librarySizeFactors(hoge)
hoge <- normalize(hoge)
plotTSNE(hoge, colour_by = "Group")
data <- logcounts(hoge)
colnames(data) <- colData(hoge)$Group
colnames(data)
data <- data[, order(colnames(data))]
colnames(data)
colnames(data) <- gsub("Group", "G", colnames(data))
colnames(data)
table(colnames(data))
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
配列長とカウント数の関係
RNA-seqデータは、原理的に配列長が長い転写物ほどその断片配列のリード数が多い傾向にあります。ここではそれを眺めます。
2014年7月3日に、boxplotで示すためにparam個で分割(20分割など)するテクニックとして「floor(nrow(data)/param)+1」としていましたが、
これだと割り切れる場合でも+1してしまうことが判明したため「ceiling(nrow(data)/param)」に修正しました(佐伯亘平氏提供情報)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。
横軸:配列長、縦軸:カウント数のシンプルな散布図を作成したい場合です。
(ダイナミックレンジが広いので両軸ともにlog10にしています。したがって、ゼロカウントになるところはlogを計算できないのでプロットされませんよ、という警告が出ます)
in_f <- "sample_length_count.txt"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
plot(data, log="xy")
#plot(Count ~ Length, data=data, log="xy")
#plot(Count ~ Length, data=log10(data), xlab="log10(Length)", ylab="log10(Count)")
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。
横軸:配列長、縦軸:カウント数のシンプルな散布図を作成したい場合です。線形回帰も行っています。
(ダイナミックレンジが広いので両軸ともにlog10にしています。したがって、ゼロカウントになるところはlogを計算できないのでプロットされませんよ、という警告が出ます)
in_f <- "sample_length_count.txt"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
head(data)
data <- data[data[,2]>0,]
dim(data)
head(data)
plot(data, log="xy")
out <- lm(Count ~ Length, data=log10(data))
abline(out, col="red")
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。
横軸:配列長、縦軸:カウント数のシンプルな散布図を作成したい場合です。平滑化曲線を追加しています。
(ダイナミックレンジが広いので両軸ともにlog10にしています。したがって、ゼロカウントになるところはlogを計算できないのでプロットされませんよ、という警告が出ます)
in_f <- "sample_length_count.txt"
param <- 0.2
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
head(data)
data <- data[data[,2]>0,]
dim(data)
head(data)
plot(data, log="xy")
lines(lowess(data, f=param), col="red")
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。
横軸:配列長、縦軸:カウント数のboxplot(箱ひげ図)を作成したい場合です。
in_f <- "sample_length_count.txt"
param_bin <- 20
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
head(data)
data <- data[data[,2]>0,]
dim(data)
head(data)
data <- data[order(data[,1]),]
head(data)
tail(data)
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
head(f)
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
data_shortest <- data[f==1,]
summary(data_shortest)
summary(log10(data_shortest))
data_longest <- data[f==param_bin,]
summary(data_longest)
summary(log10(data_longest))
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。
横軸:配列長、縦軸:カウント数のboxplot(箱ひげ図)をpng形式ファイルで保存したい場合です。
in_f <- "sample_length_count.txt"
out_f <- "hoge5.png"
param_bin <- 20
param_fig <- c(600, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[data[,2]>0,]
data <- data[order(data[,1]),]
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
dev.off()
「パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)」の出力ファイルです。
カウントデータファイルは全部で6サンプル分のデータが含まれていますが、ここでは(Ensembl Gene ID列を除く)3列目の"Pro_rep3"のカウントデータと配列長との関係を調べています。
横軸:配列長、縦軸:カウント数のboxplot(箱ひげ図)をpng形式ファイルで保存したい場合です。
in_f1 <- "srp017142_count_bowtie.txt"
in_f2 <- "srp017142_genelength.txt"
out_f <- "hoge6.png"
param_bin <- 20
param2 <- 3
param_fig <- c(600, 400)
hoge <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
hoge <- hoge[, param2]
data <- cbind(len, hoge)
data <- data[data[,2]>0,]
data <- data[order(data[,1]),]
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
dev.off()
「パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)」の出力ファイルです。
RPKMデータファイルは全部で6サンプル分のデータが含まれていますが、ここでは(Ensembl Gene ID列を除く)3列目の"Pro_rep3"のカウントデータと配列長との関係を調べています。
横軸:配列長、縦軸:カウント数のboxplot(箱ひげ図)をpng形式ファイルで保存したい場合です。配列長補正によって、偏りが軽減されていることがわかります。
in_f1 <- "srp017142_RPKM_bowtie.txt"
in_f2 <- "srp017142_genelength.txt"
out_f <- "hoge7.png"
param_bin <- 20
param2 <- 3
param_fig <- c(600, 400)
hoge <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
hoge <- hoge[, param2]
data <- cbind(len, hoge)
data <- data[data[,2]>0,]
data <- data[order(data[,1]),]
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
dev.off()
書籍 | トランスクリプトーム解析 | 2.3.6 カウントデータ取得のp92のコードを実行して得られたファイルです。
(Gene ID列を除く)1列目の配列長と2列目の"Kidney"のカウントデータとの関係を調べています。
横軸:配列長、縦軸:カウント数のboxplot(箱ひげ図)をpng形式ファイルで保存したい場合です。
in_f <- "SRA000299_ensgene.txt"
out_f <- "hoge8.png"
param_bin <- 20
param2 <- c(1, 2)
param_fig <- c(600, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[, param2]
data <- data[data[,2]>0,]
data <- data[order(data[,1]),]
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
dev.off()
「マップ後 | カウント情報取得 | paired-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)」の例題2実行結果ファイル(と同じ)です。
横軸:配列長、縦軸:カウント数のboxplot(箱ひげ図)をpng形式ファイルで保存したい場合です。
in_f1 <- "SRR616268sub_count2.txt"
in_f2 <- "SRR616268sub_genelen2.txt"
out_f <- "hoge9.png"
param_bin <- 20
param_fig <- c(600, 400)
hoge <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
data <- cbind(len, hoge)
data <- data[data[,2]>0,]
data <- data[order(data[,1]),]
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
dev.off()
「マップ後 | カウント情報取得 | paired-end | ゲノム | アノテーション無 | QuasR(Gaidatzis_2015)」の例題1実行結果ファイル(と同じ)です。
横軸:配列長、縦軸:カウント数のboxplot(箱ひげ図)をpng形式ファイルで保存したい場合です。
in_f1 <- "SRR616268sub_count1.txt"
in_f2 <- "SRR616268sub_genelen1.txt"
out_f <- "hoge10.png"
param_bin <- 20
param_fig <- c(600, 400)
hoge <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
data <- cbind(len, hoge)
data <- data[data[,2]>0,]
data <- data[order(data[,1]),]
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
dev.off()
正規化 | について
以下は記述内容が相当古いので参考程度にしてください(2014/06/22)。。。
サンプル内正規化周辺:
一つ上の項目(前処理 | 発現レベルの定量化について)は主にリファレンス配列(ゲノムやトランスクリプトーム)に対してRNA-seqによって得られたリードをマップし、
既知の遺伝子構造に対して転写物ごとの発現レベルに応じたspliced readsの割り当て方や遺伝子レベルの発現量をどのように見積もるか?といった問題に対処するプログラムを紹介しています。
そのため、サンプル内での異なる遺伝子間の発現レベルの大小を比較可能にしたうえでqPCR結果とのlinearityなどの評価を行うことになりますので、「(gene lengthなどの)サンプル内の正規化」は内包していると考えても基本的に差し支えはありません。
「長い遺伝子ほどマップされるリード数が多くなる」ことは2008年頃の時点ですでにわかっていたので、gene lengthの補正は基本的に組み込まれていました。
しかし、その後gene length補正後でも「配列長が長いほど(発現レベルが高いほど)有意に発現変動していると検出される傾向にある(Oshlack and Wakefield, 2009)」という報告や、
「GC含量が高くても低くてもリードカウント数が少なくなり、中程度のGC含量のところがリードカウント数が多い傾向にある(unimodal; Risso et al., 2011)」といったことがわかってきており、リードカウント数(or 発現変動解析結果)とGC含量(or gene length)のbiasをなくす方法が提案されてきました。
どの順番(またはタイミング)で正規化を行うかについてはまだよくわかっていません。Young(2010)らはGene Ontology解析時に発現変動解析後にgene lengthの補正を行っていますが、Risso(2011)らは発現変動解析前にGC-related bias補正を行うほうがよいと述べています。
尚、ここでの主な評価基準は下記のa.およびb.です。
サンプル間正規化周辺:
sequence depth関連のサンプル間正規化の主な目的の一つとして、異なるサンプル(グループ)間で発現の異なる遺伝子(Differentially Expressed Genes; DEGs)を検出することが挙げられます。
したがって、特にサンプル間正規化を目的とした正規化法の評価は下記のよく用いられる評価基準のうち、「c.」が重要視されます。
「どの正規化法を使うか?」ということが「どの発現変動遺伝子検出法を使うか?」ということよりも結果に与える影響が大きい(Bullard et al., 2010; Kadota et al., 2012; Garmire and Subramaniam, 2012)ことがわかりつつあります。
正規化 | サンプル間 | 複製あり | DEGES/TbT正規化(Kadota_2012)でも述べていますが、従来のサンプル間正規化法(RPMやTMM法)は比較する二群間(G1群 vs. G2群)でDEG数に大きな偏りがないことを前提にしています。
つまり、「(G1群 >> G2群)のDEG数と(G1群 << G2群)のDEG数が同程度」という前提条件がありますが、実際には大きく偏りがある場合も想定すべきで、偏りがあるなしにかかわらずロバストに正規化を行ってくれるのがTbT正規化法(Kadota et al., 2012)です。
偏りがあるなしの主な原因はDEGの分布にあるわけです。よって、TbT論文では、「正規化中にDEG検出を行い、DEGを除き、non-DEGのみで再度正規化係数を決める」というDEG elimination strategyに基づく正規化法を提案しています。
そして、TbT法を含むDEG elimination strategyに基づくいくつかの正規化法をTCCというRパッケージ中に実装しています(TCC論文中ではこの戦略をDEGES「でげす」と呼んでいます; Sun et al., 2013)。TCCパッケージ中に実装されている主な方法および入力データは以下の通りです:
- DEGES/TbT:オリジナルのTbT法そのもの。TMM-baySeq-TMMという3ステップで正規化を行う方法。step 2のDEG同定のところで計算時間のかかるbaySeqを採用しているため、他のパッケージでは数秒で終わる正規化ステップが数十分程度はかかるのがネック。また、biological replicatesがあることを前提としている。(A1, A2, ...) vs. (B1, B2, ...)のようなデータという意味。
- iDEGES/edgeR:DEGES/TbT法の超高速版(100倍程度高速)。TMM-(edgeR-TMM)nというステップで正規化を行う方法(デフォルトはn = 3)。step 2のDEG同定のところでedgeRパッケージ中のexact test (Robinson and Smyth, 2008)を採用しているためDEG同定の高速化が可能。
したがって、(step2 - step3)のサイクルをiterative (それゆえDEGESの頭にiterativeの意味を示すiがついたiDEGESとなっている)に行ってよりロバストな正規化係数を得ることが理論的に可能な方法です。それゆえreplicatesのある二群間比較を行いたい場合にはDEGES/TbTよりもiDEGES/edgeRのほうがおすすめ。
- iDEGES/DESeq:一連のmulti-stepの正規化手順をすべてDESeqパッケージ中の関数のみで実装したパイプライン。複製実験のない二群間比較時(i.e., A1 vs. B1など)にはこちらを利用します。
ここでは、主に生のリードカウント(整数値;integer)データを入力として(異なる遺伝子間の発現レベルの大小比較のため)サンプル内で行うGC含量補正やgene length補正を行う方法や、
(サンプル間で同一遺伝子の発現変動比較のため)サンプル間で行うsequence depth関連の補正を行う方法についてリストアップしておきます。なぜ前処理 | 発現レベルの定量化についてで得られた数値データを使わないのか?についてですが、
よく言われることとしては「モデル(Poisson model, generalized Poisson model, negative binomial modelなど)に従わなくなるから取り扱いずらくなる」ということもあるんでしょうね。このあたりはedgeRのマニュアルなどで明記されていたりもします。
spliced read情報を含んだ発現情報を用いた正規化法の開発についてもディスカッションではよく述べられています(Dillies et al., in press)が、、、対応版は近い将来publishされる論文で出てくるでしょう。
また、現在の方法は主に転写物レベルではなく遺伝子レベルの解析です。一歩踏み込んだエクソンレベルの解析用Rパッケージ(正規化法含む)も出てきています(Anders et al., 2012)。
一般によく用いられるカウントデータは「2塩基ミスマッチまで許容して一か所にのみマップされるリードの数(uniquely mapped reads allowing up to two mismatches)」です(Frazee et al., 2011; Risso et al., 2011; Sun and Zhu, 2012)。
どの正規化法がいいかを評価する基準としてよく用いられているのは下記のものなどが挙げられます。
二番目については、gold standardの結果自体も測定誤差(measurement error; ME)を含むのでgold standard非依存のME modelを利用した評価法(Sun and Zhu, 2012)も提案されています:
- 複製実験データの再現性が高いかどうか?(reproducibilityとかprecisionという用語が対応します)
- qRT-PCRやmicroarrayなどの他の実験技術由来のデータをgold standardとしてそれとの相関係数が高いかどうか?(accuracy; Garmire and Subramaniam, 2012)
- シミュレーションデータなど、既知の発現変動遺伝子(DEGs)を発現変動遺伝子として上位にランキングできる正規化法かどうか?(AUC; Kadota et al., 2012)
「方法名」:「Rパッケージ名」:「原著論文」の順番です。(あれば)
また、必ずしも正規化法の原著論文とは対応付いていませんのであしからず。この方法を使いたいがどのパッケージで利用可能?、という視点でご覧になってください。
- 正規化(サンプル内; GC含量補正):
-
- regression正規化(loess):EDASeq:Risso et al., BMC Bioinformatics, 2011
- global正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- full quantile (FQ)正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- conditional quantile正規化(CQN):cqn:Hansen et al., Biostatistics, 2012
- GCcorrect:Benjamini and Speed, Nucleic Acids Res., 2012
- 正規化(サンプル内; GC contents and gene length同時補正):
-
- 正規化(サンプル内; 全般;分布系):
-
- 正規化(サンプル内; 全般;その他(自分の前後の塩基の使用頻度を考慮, sequencing preference)):
-
- 正規化(サンプル間):
-
- RPM (or CPM)正規化:edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化:edgeR:Robinson et al., Bioinformatics, 2010
- Anders and Huberの(AH)正規化:DESeq:Anders and Huber, Genome Biol, 2010
- median正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- Upper-quartile (UQ)正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- full quantile (FQ)正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- TbT (DEGES/TbT)正規化(複製あり、なしデータ用):TCC:Kadota et al., Algorithms Mol. Biol., 2012
- iDEGES/edgeR正規化(複製ありデータ用):TCC:Sun et al., BMC Bioinformatics, 2013
- iDEGES/DESeq正規化(複製なしデータ用):TCC:Sun et al., BMC Bioinformatics, 2013
他の参考文献:
- an exact test for negative binomial distribution:Robinson and Smyth, Biostatistics, 2008
- Oshlack and Wakefield, Biology Direct, 2009
- goseq:Young et al., Genome Biol., 2010
- Bullard et al., BMC Bioinformatics, 2010
- ReCount:Frazee et al., BMC Bioinformatics, 2011
- Garmire and Subramanian, RNA, 2012
- DEXseq:Anders et al., Genome Res., 2012
- Sun and Zhu, Bioinformatics, 2012
- Dillies et al., Brief. Bioinform., in press
正規化 | 基礎 | RPK or CPK (配列長補正)
ここでは、遺伝子(転写物)ごとのリード数を「配列長が1000 bp (one kilobase)だったときのリード数; Reads per kilobase (RPK)」に変換するやり方を示します。
「リード数 = カウント数」なのでReadsのところをCountsに置き換えた表現(Counts per kilobase; CPK)もときどき見受けられます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。基本形です。
in_f <- "sample_length_count.txt"
out_f <- "hoge1.txt"
param <- 1000
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
nf <- param/data[,1]
out <- data[,2] * nf
tmp <- cbind(rownames(data), data[,1], out)
colnames(tmp) <- c("ID", "Length", "Count")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。
横軸:配列長、縦軸:RPK値のboxplot(箱ひげ図)をpng形式ファイルで保存するやり方です。
in_f <- "sample_length_count.txt"
out_f <- "hoge2.png"
param_bin <- 20
param_fig <- c(600, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
data <- data[data[,2]>0,]
head(data)
data <- data[order(data[,1]),]
head(data)
f <- gl(param_bin, ceiling(nrow(data)/param_bin), nrow(data))
nf <- 1000/data[,1]
data[,2] <- data[,2] * nf
head(data)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(f, log10(data[,2]), xlab="Length", ylab="log10(Count)")
abline(h=1, col="red")
dev.off()
18,110 genes×10 samplesのカウントデータファイルを一気に長さ補正するやり方です。
in_f1 <- "data_marioni.txt"
in_f2 <- "length_marioni.txt"
out_f <- "hoge3.txt"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
dim(len)
head(data)
head(len)
colSums(data)
nf <- 1000/len[,1]
data <- sweep(data, 1, nf, "*")
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「マップ後 | カウント情報取得 | paired-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)」の例題2実行結果ファイル(と同じ)です。
in_f1 <- "SRR616268sub_count2.txt"
in_f2 <- "SRR616268sub_genelen2.txt"
out_f <- "hoge4.txt"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
dim(len)
head(data, n=8)
head(len, n=8)
colSums(data)
nf <- 1000/len[,1]
data <- sweep(data, 1, nf, "*")
head(data, n=8)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
正規化 | 基礎 | RPM or CPM (総リード数補正)
カウントデータファイルを読み込んで、転写物ごとのリード数を「総リード数が100万 (million)だったときのリード数; Reads per million (RPM)」に変換するやり方を示します。
「リード数 = カウント数」なのでReadsのところをCountsに置き換えた表現(Counts per million; CPM)もときどき見受けられます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。
in_f <- "sample_length_count.txt"
out_f <- "hoge1.txt"
param1 <- 1000000
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
sum(data[,2])
nf <- param1/sum(data[,2])
out <- data[,2] * nf
head(out)
sum(out)
tmp <- cbind(rownames(data), data[,1], out)
colnames(tmp) <- c("ID", "Length", "Count")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param1 <- 1000000
param_nonDEG <- 2001:10000
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
dim(data)
colSums(data)
data <- data[param_nonDEG,]
head(data)
dim(data)
colSums(data)
nf <- param1/colSums(data)
data <- sweep(data, 2, nf, "*")
head(data)
colSums(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。non-DEGデータのみを正規化してM-A plotを作成しています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.txt"
param1 <- 1000000
param_nonDEG <- 2001:10000
param_G1 <- 3
param_G2 <- 3
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[param_nonDEG,]
nf <- param1/colSums(data)
data <- sweep(data, 2, nf, "*")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
G1 <- apply(as.matrix(data[,data.cl==1]), 1, mean)
G2 <- apply(as.matrix(data[,data.cl==2]), 1, mean)
M <- log2(G2) - log2(G1)
A <- (log2(G1) + log2(G2))/2
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
grid(col="gray", lty="dotted")
median(M, na.rm=TRUE)
abline(h=median(M, na.rm=TRUE), col="black")
tmp <- cbind(rownames(data), data, G1, G2, log2(G1), log2(G2), M, A)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。non-DEGデータのみを正規化してM-A plotを作成していろいろとハイライトさせるやり方です。
in_f <- "data_hypodata_3vs3.txt"
param1 <- 1000000
param_nonDEG <- 2001:10000
param_G1 <- 3
param_G2 <- 3
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[param_nonDEG,]
nf <- param1/colSums(data)
data <- sweep(data, 2, nf, "*")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
G1 <- apply(as.matrix(data[,data.cl==1]), 1, mean)
G2 <- apply(as.matrix(data[,data.cl==2]), 1, mean)
M <- log2(G2) - log2(G1)
A <- (log2(G1) + log2(G2))/2
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
grid(col="gray", lty="dotted")
obj <- "gene_2002"
points(A[obj], M[obj], col="red", cex=1.0, pch=20)
obj <- (M >= log2(4))
points(A[obj], M[obj], col="blue", cex=0.8, pch=6)
sum(obj, na.rm=TRUE)
obj <- (A > 10)
points(A[obj], M[obj], col="lightblue", cex=0.8, pch=17)
sum(obj, na.rm=TRUE)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。上記DEGを色分けするやり方です。
in_f <- "data_hypodata_3vs3.txt"
param1 <- 1000000
param_G1 <- 3
param_G2 <- 3
param_DEG_G1 <- 1:1800
param_DEG_G2 <- 1801:2000
param_nonDEG <- 2001:10000
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
nf <- param1/colSums(data)
data <- sweep(data, 2, nf, "*")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
G1 <- apply(as.matrix(data[,data.cl==1]), 1, mean)
G2 <- apply(as.matrix(data[,data.cl==2]), 1, mean)
M <- log2(G2) - log2(G1)
A <- (log2(G1) + log2(G2))/2
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
grid(col="gray", lty="dotted")
median(M[param_nonDEG], na.rm=TRUE)
abline(h=median(M[param_nonDEG], na.rm=TRUE), col="black")
obj <- param_DEG_G1
points(A[obj], M[obj], col="blue", cex=0.1, pch=20)
obj <- param_DEG_G2
points(A[obj], M[obj], col="red", cex=0.1, pch=20)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
上記DEGを色分けしてPNG形式ファイルとして保存するやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge6.png"
param1 <- 1000000
param_G1 <- 3
param_G2 <- 3
param_DEG_G1 <- 1:1800
param_DEG_G2 <- 1801:2000
param_nonDEG <- 2001:10000
param_fig <- c(400, 380)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
nf <- param1/colSums(data)
data <- sweep(data, 2, nf, "*")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
G1 <- apply(as.matrix(data[,data.cl==1]), 1, mean)
G2 <- apply(as.matrix(data[,data.cl==2]), 1, mean)
M <- log2(G2) - log2(G1)
A <- (log2(G1) + log2(G2))/2
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)",
ylim=c(-8, 6), pch=20, cex=.1)
grid(col="gray", lty="dotted")
median(M[param_nonDEG], na.rm=TRUE)
abline(h=median(M[param_nonDEG], na.rm=TRUE), col="black")
obj <- param_DEG_G1
points(A[obj], M[obj], col="blue", cex=0.1, pch=20)
obj <- param_DEG_G2
points(A[obj], M[obj], col="red", cex=0.1, pch=20)
legend("topright", c("DEG(G1)", "DEG(G2)", "non-DEG"), col=c("blue", "red", "black"), pch=20)
dev.off()
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
edgeRパッケージ中のcpm関数を用いるやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge7.txt"
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
colSums(data)
data <- cpm(data)
head(data)
colSums(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
param_G1 <- 3
param_G2 <- 3
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
grid(col="gray", lty="dotted")
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
6.をTCCパッケージを利用して行うやり方です。
各サンプルのリード数を100万ではなく平均リード数に揃えています。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8.png"
param_G1 <- 3
param_G2 <- 3
param_DEG_G1 <- 1:1800
param_DEG_G2 <- 1801:2000
param_nonDEG <- 2001:10000
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(data), normalized)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
obj <- c(rep(1, length(param_DEG_G1)),
rep(2, length(param_DEG_G2)),
rep(3, length(param_nonDEG)))
cols <- c("blue", "red", "black")
plot(tcc, col=cols, col.tag=obj, normalize=T)
legend("topright", c("DEG(G1)", "DEG(G2)", "non-DEG"), col=c("blue", "red", "black"), pch=20)
dev.off()
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
2.をTCCパッケージを利用して行うやり方です。
各サンプルのリード数を100万ではなく平均リード数に揃えています。non-DEGデータのみを正規化してM-A plotを作成するやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge9.txt"
out_f2 <- "hoge9.png"
param_G1 <- 3
param_G2 <- 3
param_nonDEG <- 2001:10000
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[param_nonDEG,]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(data), normalized)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, normalize=T)
dev.off()
in_f <- "sample_blekhman_36.txt"
out_f <- "hoge10.txt"
param1 <- 1000000
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
nf <- param1/colSums(data)
data <- sweep(data, 2, nf, "*")
colSums(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
正規化 | 基礎 | RPKM(Mortazavi_2008)
遺伝子(転写物)ごとのリード数を「配列長が1000 bp (kilobase)で総リード数が100万だったときのリード数;
Reads per kilobase per million (RPKM)」に変換するやり方(Mortazavi et al., 2008)を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。出力ファイルの3列目がRPKMに変更されています。
in_f <- "sample_length_count.txt"
out_f <- "hoge1.txt"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
sum(data[,2])
nf_RPM <- 1000000/sum(data[,2])
nf_RPK <- 1000/data[,1]
data[,2] <- data[,2] * nf_RPM * nf_RPK
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
18,110 genes×10 samples (G1群5サンプル vs. G2群5サンプル)のカウントデータファイルと長さ情報ファイルです。
in_f1 <- "data_marioni.txt"
in_f2 <- "length_marioni.txt"
out_f <- "hoge2.txt"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
dim(len)
head(data)
head(len)
colSums(data)
nf_RPM <- 1000000/colSums(data)
data <- sweep(data, 2, nf_RPM, "*")
head(data)
colSums(data)
nf_RPK <- 1000/len[,1]
data <- sweep(data, 1, nf_RPK, "*")
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「マップ後 | カウント情報取得 | paired-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)」の例題2実行結果ファイル(と同じ)です。
in_f1 <- "SRR616268sub_count2.txt"
in_f2 <- "SRR616268sub_genelen2.txt"
out_f <- "hoge3.txt"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
dim(len)
head(data)
head(len)
colSums(data)
nf_RPM <- 1000000/colSums(data)
data <- sweep(data, 2, nf_RPM, "*")
head(data)
colSums(data)
nf_RPK <- 1000/len[,1]
data <- sweep(data, 1, nf_RPK, "*")
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題1と同じですが、TPM (Transcripts Per Million)
(Li et al., Bioinformatics, 2010)の計算手順との違いがわかるような書き方にしています。
in_f <- "sample_length_count.txt"
out_f <- "hoge4.txt"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
sum(data[,2])
nf_RPM <- 1000000/sum(data[,2])
RPM <- data[,2] * nf_RPM
nf_RPK <- 1000/data[,1]
RPKM <- RPM * nf_RPK
head(RPKM)
tmp <- cbind(rownames(data), data[,1], RPKM)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
正規化 | 基礎 | TPM(Li_2010)
TPM (Transcripts Per Million) (Li et al., Bioinformatics, 2010)補正を行うやり方を示します。
RPKMは理論上RPM補正とRPK補正を同時に行っていますが、TPM補正は先にRPK補正をやってから"RPK補正後のデータを100万(one million)に揃える"やり方です。
補正後の数値の総和は、RPKMだとサンプル間で一致しないのに対して、TPMだと一致するというメリットがあります。
(サンプルごとのRPKM補正後の数値の総和を100万に揃えたものが、TPMという理解でよいはずです。)
ちなみにTranscripts Per Millionの略としてTPMになっていますが、実際にはkilobaseに揃える配列長補正も行っています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1-3列目がそれぞれ、gene ID, 配列長, カウント数からなるファイルです。出力ファイルの3列目がTPMに変更されています。
in_f <- "sample_length_count.txt"
out_f <- "hoge1.txt"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
sum(data[,2])
nf_RPK <- 1000/data[,1]
RPK <- data[,2] * nf_RPK
head(RPK)
nf_RPM <- 1000000/sum(RPK)
TPM <- RPK * nf_RPM
head(TPM)
tmp <- cbind(rownames(data), data[,1], TPM)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
正規化 | サンプル内 | EDASeq(Risso_2011)
Risso et al., BMC Bioinformatics, 2011のFig.1を見ればわかりますが、
遺伝子のカウント数(発現量に相当;縦軸)をGC含量で横軸に展開してやるとunimodal(山が一つ、の意味)な分布になる傾向にあります。
つまり、GC含量が低すぎても高すぎてもカウント数が低くなり、中程度のときにタグカウント数が多くなる傾向にある、ということを言っているわけです。
彼らはこの傾向をYeastのRNA-seqデータ(同じgrowth conditionで得られた4つのcultures。これらはbiological replicatesとみなせるそうです。
そしてcultureごとに2 technical replicatesがある。計8列分のデータ; Y1, Y1, Y2, Y2, Y7, Y7, Y4, Y4; SRA048710)で示しています。
このデータから4 replicates vs. 4 replicatesの"null pseudo-dateset"を計choose(8,4)/2 = 35セットの「発現変動遺伝子(DEG)のないデータセット」を作成可能ですが、
GC含量が高いほどDEGとcallされる傾向にある(Fig.6)ことも示しています。
マイクロアレイではこのようなGC biasは見られませんので、library preparation時にこのようなbiasが導入されているんだろう(Benjamini and Speed, Nucleic Acids Res., 2012)と言われています。
Rissoらは、このGC biasを補正するための方法として、EDASeqパッケージ中でloess, median, FQ (full quantile)の三つの正規化法を提案し実装していますが、ここでは彼らの論文中で最もGC biasを補正できたというFQ正規化法のやり方を示します。
もちろんGC biasを補正するわけですから、入力データはいわゆる「遺伝子発現行列データ(count matrix)」以外に「遺伝子ごとのGC含量情報を含むベクトル情報」が必要です。
ここではサンプルデータ10のyeastRNASeqパッケージから取得可能な「2 samples vs 2 samples」のyeast RNA-seq dataset (7065 genes×4列; Lee et al., 2008)、
およびEDASeqパッケージから取得可能なSGD ver. r64に基づくyeast遺伝子のGC含量情報(6717 genes)を用いて一連の解析を行います。
発現情報とGC含量情報の対応付けが若干ややこしいですが、このあたりのテクニックは結構重要です。
尚、以下ではparam1のところで、"full"を指定することでfull quantile正規化(GCのbinごとにカウントの順位が同じものは同じカウントになるような正規化)を実現していますが、ここを以下のように変えることで他に三種類の異なる正規化を実行することができます:
- "loess"の場合:loess正規化(non-linearな局所的な回帰を行ってくれる)
- "median"の場合:median正規化(GCのbinごとのmedianが同じになるような正規化を行う)
- "upper"の場合:upper quartile正規化(GCのbinごとのupper quartile (75 percentile rankのこと)が同じになるような正規化を行う)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてある(or 出力ファイルを置きたい)ディレクトリに移動し以下をコピペ。
in_f <- "sample_gc_count.txt"
out_f <- "hoge1.txt"
param1 <- "full"
param2 <- 10
library(EDASeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
count <- as.matrix(data[,2])
rownames(count) <- rownames(data)
GC <- data.frame(GC=data[,1]/100)
rownames(GC) <- rownames(data)
head(count)
head(GC)
data.cl <- c(rep(1, ncol(count)))
es <- newSeqExpressionSet(exprs=count,
featureData=GC,
phenoData=data.frame(conditions=data.cl))
es
out <- withinLaneNormalization(es, "GC", which=param1, num.bins=param2, round=FALSE)
tmp <- cbind(rownames(data), GC, exprs(out))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
(どういう正規化係数がかかっているのかについて詳細な情報を出力している)
in_f <- "sample_gc_count.txt"
out_f1 <- "hoge2_before.png"
out_f2 <- "hoge2_after.png"
out_f3 <- "hoge2.txt"
param1 <- "full"
param2 <- 10
param_fig <- c(500, 450)
library(EDASeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
count <- as.matrix(data[,2])
rownames(count) <- rownames(data)
GC <- data.frame(GC=data[,1]/100)
rownames(GC) <- rownames(data)
head(count)
head(GC)
data.cl <- c(rep(1, ncol(count)))
es <- newSeqExpressionSet(exprs=count,
featureData=GC,
phenoData=data.frame(conditions=data.cl))
es
out <- withinLaneNormalization(es, "GC", which=param1, num.bins=param2, round=FALSE)
out2 <- withinLaneNormalization(es, "GC", which=param1, num.bins=param2, offset=TRUE)
png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])
biasPlot(es, "GC", log = T)
dev.off()
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
biasPlot(out, "GC", log = T)
dev.off()
tmp <- cbind(rownames(data), data, exp(offst(out2)), exprs(out))
colnames(tmp) <- c("ID", "GC_contents", "Count(before)", "Norm_factor", "Count(after")
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
3. EDASeqパッケージ中の記述と似たやり方で示した場合:
param_G1 <- 2
param_G2 <- 2
param1 <- "full"
library(yeastRNASeq)
library(EDASeq)
data(geneLevelData)
data(yeastGC)
dim(geneLevelData)
head(geneLevelData)
length(yeastGC)
head(yeastGC)
head(rownames(geneLevelData))
head(names(yeastGC))
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
common <- intersect(rownames(geneLevelData), names(yeastGC))
length(common)
data <- as.matrix(geneLevelData[common, ])
GC <- data.frame(GC = yeastGC[common])
head(rownames(data))
head(rownames(GC))
es <- newSeqExpressionSet(exprs = data,
featureData = GC,
phenoData = data.frame(conditions = data.cl,
row.names = colnames(data)))
es
biasPlot(es, "GC", log = T, ylim = c(-1, 4))
out <- withinLaneNormalization(es, "GC", which = param1)
biasPlot(out, "GC", log = T, ylim = c(-1, 4))
out_f1 <- "data_yeastGCbias_common_before.txt"
out_f2 <- "data_yeastGCbias_common_after.txt"
out_f3 <- "data_yeastGCbias_common_GCcontent.txt"
tmp <- cbind(rownames(exprs(es)), exprs(es))
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(exprs(out)), exprs(out))
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(fData(es)), fData(es))
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
out_f4 <- "data_yeastGCbias_common_offset.txt"
out2 <- withinLaneNormalization(es, "GC", which = param1, offset = TRUE)
head(offst(out2))
tmp <- cbind(rownames(offst(out2)), offst(out2))
write.table(tmp, out_f4, sep="\t", append=F, quote=F, row.names=F)
head(exprs(out))
head(exprs(es))
head(exp(offst(out2)))
normalized <- exprs(es) * exp(offst(out2))
head(normalized)
head(round(normalized))
4. 二つのタブ区切りテキストファイルの読み込みからやる場合:
7,065行×4列のyeast RNA-seqデータ(data_yeast_7065.txt; 2 wild-types vs. 2 mutant strains; technical replicates)
6,717 yeast genes (SGD ver. r64)のGC含量(yeastGC_6717.txt)
in_f1 <- "data_yeast_7065.txt"
in_f2 <- "yeastGC_6717.txt"
param_G1 <- 2
param_G2 <- 2
param1 <- "full"
library(EDASeq)
data.tmp <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
gc.tmp <- read.table(in_f2, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
common <- intersect(rownames(data.tmp), rownames(gc.tmp))
length(common)
data <- as.matrix(data.tmp[common, ])
GC <- data.frame(GC = gc.tmp[common, ])
rownames(GC) <- common
es <- newSeqExpressionSet(exprs = data,
featureData = GC,
phenoData = data.frame(conditions = data.cl,
row.names = colnames(data)))
es
biasPlot(es, "GC", log = T, ylim = c(-1, 4))
out <- withinLaneNormalization(es, "GC", which = param1)
biasPlot(out, "GC", log = T, ylim = c(-1, 4))
out_f1 <- "data_yeastGCbias_common_before.txt"
out_f2 <- "data_yeastGCbias_common_after.txt"
out_f3 <- "data_yeastGCbias_common_GCcontent.txt"
tmp <- cbind(rownames(exprs(es)), exprs(es))
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(exprs(out)), exprs(out))
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(fData(es)), fData(es))
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
正規化 | サンプル内 | RNASeqBias(Zheng_2011)
RNASeqBiasというRパッケージの正規化法の論文です。論文中では、GAM correction法と呼んでいます。
a simple generalized-additive-model based approachを用いてGC contentやgene lengthの補正を同時に行ってくれるようです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
正規化 | サンプル間 | について
RNA-seqのサンプル間正規化法をまとめます。scRNA-seq用のプログラムは、2019年09月27日に
「正規化 | scRNA-seq | について」に移動させました。
R用:
- RPM (or CPM)正規化:edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化:edgeR:Robinson et al., Bioinformatics, 2010
- Anders and Huberの(AH)正規化:DESeq:Anders and Huber, Genome Biol, 2010
- PoissonSeqの正規化(nonDEGを使う発想自体はDEGESと同じ):PoissonSeq:Li et al., Biostatistics, 2012
- median正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- Upper-quartile (UQ)正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- full quantile (FQ)正規化:EDASeq:Risso et al., BMC Bioinformatics, 2011
- TbT (DEGES/TbT)正規化(複製あり、なしデータ用):TCC:Kadota et al., Algorithms Mol. Biol., 2012
- iDEGES/edgeR正規化(複製ありデータ用):TCC:Sun et al., BMC Bioinformatics, 2013
- iDEGES/DESeq正規化(複製なしデータ用):TCC:Sun et al., BMC Bioinformatics, 2013
- RUV正規化(spike-insの利用を推奨):Risso et al., Nat Biotechnol., 2014
- GP-Theta:deGPS:Chu et al., BMC Genomics, 2015
- MRN正規化:Maza et al., Commun Integr Biol., 2013
- RUV正規化を実装したパッケージ:RUVSeq:Peixoto et al., Nucleic Acids Res., 2015
正規化 | サンプル間 | Upper-quartile(Bullard_2010)
Bullardら(2010)の各種統計的解析手法の評価論文で用いられた正規化法です。
マイクロアレイデータのときは「中央値(median)を揃える」などが行われたりしましたが、
「第3四分位数(upper-quartile or 75 percentile rank)の値を揃える」という操作を行うことに相当します。
EDASeqというパッケージを用いて正規化を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "data_hypodata_3vs3_UQ.txt"
param_G1 <- 3
param_G2 <- 3
param1 <- "upper"
library(EDASeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
es <- newSeqExpressionSet(exprs = as.matrix(data),
phenoData = data.frame(conditions = data.cl,
row.names = colnames(data)))
es
summary(exprs(es))
hoge <- betweenLaneNormalization(es, which=param1)
normalized.count <- exprs(hoge)
summary(normalized.count)
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
nonDEG <- 2001:10000
boxplot(log(normalized.count[nonDEG, ]))
summary(normalized.count[nonDEG, ])
apply(normalized.count[nonDEG, ], 2, median)
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
in_f <- "data_yeast_7065.txt"
out_f <- "data_yeast_7065_UQ.txt"
param_G1 <- 2
param_G2 <- 2
param1 <- "upper"
library(EDASeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
es <- newSeqExpressionSet(exprs = as.matrix(data),
phenoData = data.frame(conditions = data.cl,
row.names = colnames(data)))
es
summary(exprs(es))
hoge <- betweenLaneNormalization(es, which=param1)
normalized.count <- exprs(hoge)
summary(normalized.count)
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
正規化 | サンプル間 | Quantile(Bullard_2010)
カウントデータの「サンプル(列)間でカウント数の順位が同じならばカウント数も同じ」になるような操作を行う正規化です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
library(limma)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
hoge <- normalizeBetweenArrays(as.matrix(data))
normalized.count <- exprs(hoge)
summary(normalized.count)
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
箱ひげ図(box plot)も作成しています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
library(EDASeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
es <- newSeqExpressionSet(exprs=as.matrix(data),
phenoData=data.frame(conditions=data.cl,
ow.names=colnames(data)))
es
summary(exprs(es))
hoge <- betweenLaneNormalization(es, which="full")
normalized.count <- exprs(hoge)
summary(normalized.count)
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
nonDEG <- 2001:10000
boxplot(log(normalized.count[nonDEG, ]))
summary(normalized.count[nonDEG, ])
apply(normalized.count[nonDEG, ], 2, median)
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge3.txt"
param_G1 <- 3
param_G2 <- 3
library(EDASeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
es <- newSeqExpressionSet(exprs=as.matrix(data),
phenoData=data.frame(conditions=data.cl,
ow.names=colnames(data)))
es
summary(exprs(es))
hoge <- betweenLaneNormalization(es, which="full")
normalized.count <- exprs(hoge)
summary(normalized.count)
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
正規化 | サンプル間 | 2群間 | 複製あり | iDEGES/edgeR(Sun_2013)
TCCパッケージ(Sun et al., BMC Bioinformatics, 2013)から利用可能なiDEGES/edgeR正規化法の実行手順を示します。
iDEGES/edgeRは、TbT正規化法(Kadota et al., Algorithms Mol. Biol., 2012)
以上の理論性能かつ100倍程度高速(デフォルトの場合)化を実現したお勧めの正規化法です。
ここでは、iDEGES/edgeR法の実体である「TMM-(edgeR-TMM)n」パイプラインのnの値(iterationの回数)を3として実行します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
head(normalized)
tmp <- cbind(rownames(data), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
M-A plotを作成したり、non-DEGの分布についても確認しています。
in_f <- "data_hypodata_3vs3.txt"
param_G1 <- 3
param_G2 <- 3
param_DEG_G1 <- 1:1800
param_DEG_G2 <- 1801:2000
param_nonDEG <- 2001:10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
data <- getNormalizedData(tcc)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
G1 <- apply(as.matrix(data[,data.cl==1]), 1, mean)
G2 <- apply(as.matrix(data[,data.cl==2]), 1, mean)
M <- log2(G2) - log2(G1)
A <- (log2(G1) + log2(G2))/2
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
grid(col="gray", lty="dotted")
median(M[param_nonDEG], na.rm=TRUE)
abline(h=median(M[param_nonDEG], na.rm=TRUE), col="black")
obj <- param_DEG_G1
points(A[obj], M[obj], col="blue", cex=0.1, pch=20)
obj <- param_DEG_G2
points(A[obj], M[obj], col="red", cex=0.1, pch=20)
legend("topright", c("DEG(G1)", "DEG(G2)", "non-DEG"), col=c("blue", "red", "black"), pch=20)
summary(data[param_nonDEG,])
apply(data[param_nonDEG,], 2, median)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
M-A plotをpng形式ファイルで保存するやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.png"
param_G1 <- 3
param_G2 <- 3
param_DEG_G1 <- 1:1800
param_DEG_G2 <- 1801:2000
param_nonDEG <- 2001:10000
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
data <- getNormalizedData(tcc)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
G1 <- apply(as.matrix(data[,data.cl==1]), 1, mean)
G2 <- apply(as.matrix(data[,data.cl==2]), 1, mean)
M <- log2(G2) - log2(G1)
A <- (log2(G1) + log2(G2))/2
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)",
ylim=c(-8, 6), pch=20, cex=.1)
grid(col="gray", lty="dotted")
median(M[param_nonDEG], na.rm=TRUE)
abline(h=median(M[param_nonDEG], na.rm=TRUE), col="black")
obj <- param_DEG_G1
points(A[obj], M[obj], col="blue", cex=0.1, pch=20)
obj <- param_DEG_G2
points(A[obj], M[obj], col="red", cex=0.1, pch=20)
legend("topright", c("DEG(G1)", "DEG(G2)", "non-DEG"), col=c("blue", "red", "black"), pch=20)
dev.off()
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
TCCパッケージ中のplot関数を使ってシンプルにM-A plotをpng形式ファイルで保存するやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge4.png"
param_G1 <- 3
param_G2 <- 3
param_DEG_G1 <- 1:1800
param_DEG_G2 <- 1801:2000
param_nonDEG <- 2001:10000
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
obj <- c(rep(1, length(param_DEG_G1)),
rep(2, length(param_DEG_G2)),
rep(3, length(param_nonDEG)))
cols <- c("blue", "red", "black")
plot(tcc, col=cols, col.tag=obj, ylim=c(-8, 6), median.lines=TRUE)
legend("topright", c("DEG(G1)", "DEG(G2)", "non-DEG"), col=c("blue", "red", "black"), pch=20)
dev.off()
- Robinson and Smyth, Biostatistics, 2008
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR:Robinson et al., Bioinformatics, 2010
- baySeq:Hardcastle and Kelly, BMC Bioinformatics, 2010
- TCC:Sun et al., BMC Bioinformatics, 2013
正規化 | サンプル間 | 2群間 | 複製あり | DEGES/TbT(Kadota_2012)
TbT法の正規化における基本戦略は「正規化時に悪さをするDEGを正規化前に除いてしまえ (DEG elimination strategy; DEGES; でげす)」です。
通常のDEG検出手順は「1.データ正規化( → 2.DEG検出)」の2ステップで完結するため、上記コンセプトを実現することができません。そこで通常の手順を二回繰返す解析パイプラインを提案しています。
つまり「1.データ正規化 → 2.DEG検出 → 3.データ正規化( → 4.DEG検出)」です。この手順で行えばStep3のデータ正規化時にStep2で検出されたDEG候補を除いたデータで正規化を行うことができるのです。
TbT法の実体は、「1.TMM法(Robinson and Oshlack, 2010) → 2.baySeq (Hardcastle and Kelly, 2010) → 3.TMM法」の解析パイプラインです。Step1で既存正規化法の中で高性能なTMM法を採用し、
Step2で(Step1で得られたTMM正規化係数と総リード数(library sizes)を掛けて得られた)effective library sizesを与えてbaySeqを実行してDEG候補を取得し、
Step3でnon-DEGsのみで再度TMM正規化を実行して得られたものがTbT正規化係数です。従って、TbT正規化法を利用する場合は、内部的に用いられた文献も引用するようにお願いします。
以下のスクリプトを実行すると概ね数十分程度はかかります(*_*)...。これは、TbT法の内部で計算時間のかかるbaySeqを利用しているためです。
動作確認のみしたい、という人はparam_samplesizeのところで指定する数値を500とかにして実行してください。おそらく数分で結果を得られると思われます。
ここでは、TbT正規化係数の算出法とTbT正規化後のデータを得る手順などを示しています。
が、基本的に正規化だけで終わることはありませんので、ここの項目のみでは何の役にも立ちませんのであしからず。。。
また、サンプルデータ14 (data_hypodata_1vs1.txt)の1 sample vs. 1 sampleのような「複製なし」データの正規化は実行できません(理由はstep2で用いるbaySeqが複製ありを前提としているためです)のでご注意ください。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_samplesize <- 10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm",
test.method="bayseq", samplesize=param_samplesize)
normalized <- getNormalizedData(tcc)
head(normalized)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
nonDEG <- 2001:10000
boxplot(log(normalized[nonDEG, ]))
summary(normalized[nonDEG, ])
apply(normalized[nonDEG, ], 2, median)
table(tcc$private$DEGES.potentialDEG)
tcc$norm.factors
tcc$private$simulation <- TRUE
tcc$simulation$trueDEG <- c(rep(1, 1800), rep(2, 200), rep(0, 8000))
plot(tcc, median.lines=TRUE)
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge2.txt"
param_G1 <- 2
param_G2 <- 2
param_samplesize <- 10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm",
test.method="bayseq", samplesize=param_samplesize)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
table(tcc$private$DEGES.potentialDEG)
tcc$norm.factors
このデータはどのサンプルでも発現していない(zero count; ゼロカウント)ものが多いので、
どこかのサンプルで0より大きいカウントのもののみからなるサブセットを抽出して2.と同様の計算を行っています。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 2
param_samplesize <- 10000
param_lowcount <- 0
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(tcc$count)
tcc <- filterLowCountGenes(tcc, low.count = param_lowcount)
dim(tcc$count)
tcc <- calcNormFactors(tcc, norm.method="tmm",
test.method="bayseq", samplesize=param_samplesize)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
table(tcc$private$DEGES.potentialDEG)
tcc$norm.factors
- Marioni et al., Genome Res., 2008
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- TbT正規化法:Kadota et al., Algorithms Mol. Biol., 2012
- edgeR:Robinson et al., Bioinformatics, 2010
- baySeq:Hardcastle and Kelly, BMC Bioinformatics, 2010
- TCC:Sun et al., BMC Bioinformatics, 2013
正規化 | サンプル間 | 2群間 | 複製あり | TMM(Robinson_2010)
前処理 | についてでも述べていますがNGSデータはマイクロアレイに比べてダイナミックレンジが広いという利点はあるとは思いますが、
RPM(やRPKM)で実装されているいわゆるグローバル正規化に基づく方法はごく少数の高発現遺伝子の発現レベルの影響をもろに受けます。
そしてこれらが比較するサンプル間で発現変動している場合には結論が大きく変わってしまいます。なぜなら総リード数に占める高発現の発現変動遺伝子(Differentially Expressed Genes; DEGs)のリード数の割合が大きいからです。
RobinsonらはMarioniら(2008)の「腎臓 vs. 肝臓」データの比較において実際にこのような現象が起きていることを(housekeeping遺伝子の分布を真として)示し、
少数の高発現遺伝子の影響を排除するためにtrimmed mean of M values (TMM)という正規化法を提案しています(Robinson and Oshlack, 2010)。
この方法はRのedgeRというパッケージ中にcalcNormFactorsという名前の関数で存在します。
また、この方法はTCCパッケージ中の関数を用いても実行可能です。
TCCパッケージから得られる(TMM)正規化係数は「正規化係数の平均が1になるようにさらに正規化したもの」であるため、両者の正規化係数に若干の違いがありますが細かいことは気にする必要はありません。
ここでは、正規化係数の算出と正規化後のデータを得る手順などを示していますが、基本的に正規化だけで終わることはありませんので、ここの項目のみでは何の役にも立ちませんのであしからず。。。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
nonDEG <- 2001:10000
boxplot(log(normalized[nonDEG, ]))
summary(normalized[nonDEG, ])
apply(normalized[nonDEG, ], 2, median)
tcc$norm.factors
tcc$private$simulation <- TRUE
tcc$simulation$trueDEG <- c(rep(1, 1800), rep(2, 200), rep(0, 8000))
plot(tcc, median.lines=TRUE)
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge2.txt"
param_G1 <- 2
param_G2 <- 2
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
tcc$norm.factors
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
TCCを使わずにedgeRパッケージ内の関数を用いて2.と同じ結果を出すやり方です。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 2
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
d <- DGEList(counts=data, group=data.cl)
d <- calcNormFactors(d)
d$samples$norm.factors
mean(d$samples$norm.factors)
norm.factors <- d$samples$norm.factors/mean(d$samples$norm.factors)
norm.factors
ef.libsizes <- colSums(data)*norm.factors
normalized.count <- sweep(data, 2, mean(ef.libsizes)/ef.libsizes, "*")
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
正規化 | サンプル間 | 2群間 | 複製あり | median(Anders_2010)
DESeqというRパッケージ中で採用されている正規化法を採用して得られた正規化後のデータを得るやり方を示します。
DESeqは正規化係数(normalization factor)という言葉を使わずにsize factorという言葉を使っています。
これはTbTやTMM正規化係数とは異なるものなので、ここでは「DESeqの正規化係数」を得ています。
また、この方法はTCCパッケージ中の関数を用いても実行可能です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
nonDEG <- 2001:10000
boxplot(log(normalized[nonDEG, ]))
summary(normalized[nonDEG, ])
apply(normalized[nonDEG, ], 2, median)
tcc$norm.factors
tcc$private$simulation <- TRUE
tcc$simulation$trueDEG <- c(rep(1, 1800), rep(2, 200), rep(0, 8000))
plot(tcc, median.lines=TRUE)
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge2.txt"
param_G1 <- 2
param_G2 <- 2
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
tcc$norm.factors
summary(normalized)
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
TCCを使わずにDESeqパッケージ内の関数を用いて2.と同じ結果を出すやり方です。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 2
library(DESeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
cds <- newCountDataSet(data, data.cl)
cds <- estimateSizeFactors(cds)
sizeFactors(cds)
norm.factors <- sizeFactors(cds)/colSums(data)
norm.factors <- norm.factors/mean(norm.factors)
norm.factors
sizeFactors(cds) <- sizeFactors(cds)/mean(sizeFactors(cds))
normalized.count <- counts(cds, normalized=TRUE)
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
summary(normalized.count)
technical replicatesのデータ(mut群2サンプル vs. wt群2サンプル)です。
DESeqパッケージ内の関数を用いてDESeqパッケージ内のマニュアル通りにやった場合。
若干数値が違ってきます(ということを示したいだけです)が正規化後の値の要約統計量をどこに揃えるか程度の違いなので気にする必要はないです。
実際、ここで得られるsize factorsの平均は1.019042ですが、この定数値を正規化後のデータに掛けるとTCCで得られるデータと同じになります。
in_f <- "data_yeast_7065.txt"
out_f <- "hoge4.txt"
param_G1 <- 2
param_G2 <- 2
library(DESeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
cds <- newCountDataSet(data, data.cl)
cds <- estimateSizeFactors(cds)
normalized.count <- counts(cds, normalized=TRUE)
tmp <- cbind(rownames(normalized.count), normalized.count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
summary(normalized.count)
正規化 | サンプル間 | 2群間 | 複製なし | iDEGES/DESeq(Sun_2013)
iDEGES/edgeR法(Sun et al., BMC Bioinformatics, 2013)は、サンプルデータ14 (data_hypodata_1vs1.txt)の1 sample vs. 1 sampleのような「複製なし」データの正規化は実行できません。
理由はstep2で用いるexact test(iDEGES/edgeR法の場合)が複製ありを前提としているためです。
iDEGES/DESeq法(Sun et al., BMC Bioinformatics, 2013)は複製なしの二群間比較用データの正規化を「正規化時に悪さをするDEGを正規化前に除いてしまえ (DEG elimination strategy; DEGES; でげす)」というTbT法論文(Kadota et al., 2012)で提唱した戦略を
DESeqパッケージ中の関数(正規化法やDEG検出法)のみで実現したDESeq-(DESeq-DESeq)n(デフォルトはn=3)からなるパイプラインです。
ここでは、iDEGES/DESeq正規化係数の算出法とiDEGES/DESeq正規化後のデータを得る手順などを示しています。
が、基本的に正規化だけで終わることはありませんので、ここの項目のみでは何の役にも立ちませんのであしからず。。。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, iteration=3)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
nonDEG <- 2001:10000
boxplot(log(normalized[nonDEG, ]))
summary(normalized[nonDEG, ])
apply(normalized[nonDEG, ], 2, median)
table(tcc$private$DEGES.potentialDEG)
tcc$norm.factors
tcc$private$simulation <- TRUE
tcc$simulation$trueDEG <- c(rep(1, 1800), rep(2, 200), rep(0, 8000))
plot(tcc, median.lines=TRUE)
正規化 | サンプル間 | 2群間 | 複製なし | TMM(Robinson_2010)
TMM正規化法をTCCパッケージを用いて行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
nonDEG <- 2001:10000
boxplot(log(normalized[nonDEG, ]))
summary(normalized[nonDEG, ])
apply(normalized[nonDEG, ], 2, median)
tcc$norm.factors
tcc$private$simulation <- TRUE
tcc$simulation$trueDEG <- c(rep(1, 1800), rep(2, 200), rep(0, 8000))
plot(tcc, median.lines=TRUE)
正規化 | サンプル間 | 2群間 | 複製なし | median(Anders_2010)
DESeqというRパッケージ中で採用されている正規化法を採用して得られた正規化後のデータを得るやり方を示します。
DESeqは正規化係数(normalization factor)という言葉を使わずにsize factorという言葉を使っています。
これはTbTやTMM正規化係数とは異なるものなので、ここでは「DESeqの正規化係数」を得ています。
また、この方法はTCCパッケージ中の関数を用いても実行可能です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(normalized), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc)
nonDEG <- 2001:10000
boxplot(log(normalized[nonDEG, ]))
summary(normalized[nonDEG, ])
apply(normalized[nonDEG, ], 2, median)
tcc$norm.factors
tcc$private$simulation <- TRUE
tcc$simulation$trueDEG <- c(rep(1, 1800), rep(2, 200), rep(0, 8000))
plot(tcc, median.lines=TRUE)
正規化 | サンプル間 | 3群間 | 複製あり | iDEGES/edgeR(Sun_2013)
TCCパッケージ(Sun et al., BMC Bioinformatics, 2013)から利用可能なiDEGES/edgeR正規化法の実行手順を示します。
ここでは、iDEGES/edgeR法の実体である「TMM-(edgeR-TMM)n」パイプラインのnの値(iterationの回数)を3として実行します。RPKMのような長さ補正は行っていませんのでご注意ください。ここの出力結果はRPMに相当するものです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で高発現、gene_2101〜gene_2700がG2群で高発現、gene_2701〜gene_3000がG3群で高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
head(normalized)
tmp <- cbind(rownames(data), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で高発現、gene_2101〜gene_2700がG2群で高発現、gene_2701〜gene_3000がG3群で高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
non-DEGの分布について確認しています。
in_f <- "data_hypodata_3vs3vs3.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_nonDEG <- 3001:10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
head(normalized)
summary(normalized[param_nonDEG,])
apply(normalized[param_nonDEG,], 2, median)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
head(normalized)
tmp <- cbind(rownames(data), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
non-DEGの分布について確認しています。
in_f <- "data_hypodata_2vs4vs3.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_nonDEG <- 3001:10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
head(normalized)
summary(normalized[param_nonDEG,])
apply(normalized[param_nonDEG,], 2, median)
正規化 | サンプル間 | 3群間 | 複製あり | TMM(Robinson_2010)
edgeRパッケージから利用可能なTMM正規化法(Robinson and Oshlack, Genome Biol., 2010)の実行手順を示します。RPKMのような長さ補正は行っていませんのでご注意ください。ここの出力結果はRPMに相当するものです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で高発現、gene_2101〜gene_2700がG2群で高発現、gene_2701〜gene_3000がG3群で高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
head(normalized)
tmp <- cbind(rownames(data), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で高発現、gene_2101〜gene_2700がG2群で高発現、gene_2701〜gene_3000がG3群で高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
non-DEGの分布について確認しています。
in_f <- "data_hypodata_3vs3vs3.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_nonDEG <- 3001:10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
head(normalized)
summary(normalized[param_nonDEG,])
apply(normalized[param_nonDEG,], 2, median)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
head(normalized)
tmp <- cbind(rownames(data), normalized)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
non-DEGの分布について確認しています。
in_f <- "data_hypodata_2vs4vs3.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_nonDEG <- 3001:10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
head(normalized)
summary(normalized[param_nonDEG,])
apply(normalized[param_nonDEG,], 2, median)
正規化 | scRNA-seq | について
bulk RNA-seq用は、「正規化 | サンプル間 | について」をご覧ください。
バッチ効果の補正に特化したもの?!は「batch correction用」としています。
Luecken and Theis, Mol Syst Biol., 2019の論文をベースに情報を追加していますので、
この著者らの思想のバイアスは多少かかっていると思います。
kBETの論文中でも、batch correctionでは
scranの性能は高いという報告のようですね。
とりあえず思い付きで正規化の項目を追加してはいますが、「解析 | 前処理 | scRNA-seq | について」もご覧ください。
R用:
- deconvolution(これの改良版がscranに組み込まれているとのこと):Lun et al., Genome Biol., 2016
- scran:Lun et al., F1000Res., 2016
- SCnorm:Bacher et al., Nat Methods, 2017
- kBET(batch correction用):Büttner et al., Nat Methods, 2019
- NormExpression:Wu et al., Front Genet., 2019
解析 | 一般 | アラインメント | ペアワイズ | について
ペアワイズアラインメント(pair-wise alignment)用プログラムを示します。一応ペアワイズアラインメントの枠組みに含まれると思うので、
ドットプロット(dot plot; dotplot)用のプログラムも示します。
R以外:
- JDotter(dotplot用):Brodie et al., Bioinformatics, 2004
- GATA(内部でBLASTを使用):Nix and Eisen, BMC Bioinformatics, 2005
- LASTZ:Harris RS, Ph.D. thesis, 2007
- Gepard(dotplot用):Krumsiek et al., Bioinformatics, 2007
- D-GENIES(dotplot用):Cabanettes and Klopp, PeerJ., 2018
解析 | 一般 | アラインメント | ペアワイズ | 基礎1 | Biostrings
グローバルアラインメント(global alignment;EMBOSSのneedleに相当)や
ローカルアラインメント(local alignment; EMBOSSのwaterに相当)をやってくれます。
ここでは、3つの塩基配列(seq1, seq2, and seq3)からなるmulti-FASTA形式のファイルtest2.fastaや、
通常のsingle-FASTA形式の2つのファイル(seq2.fastaとseq3.fasta)の比較を例題とします。
ちなみにこのページでは、「配列A vs. 配列B」という表記法で、配列Bに相当するほうが"subject"で配列Aに相当するほうが"pattern"です。
また、置換行列(nucleotideSubstitutionMatrix)を指定してやる必要がありますが、ここではEMBOSSの塩基配列比較時にデフォルトで用いている「EDNAFULL」という置換行列を用います。
local alignmentの結果は極めてEMBOSSのものと似ていますが(EMBOSSのスコア=34, Rのスコア=33)、global alignmentの結果は相当違っていることは認識しています(2010/6/8現在)。
おそらくギャップペナルティを計算する際の数式の違いによるものだろうと楽観していますが、もし間違っていればご指摘よろしくお願いします。
BLOSUM62やPAM250などの代表的な置換行列もftp://ftp.ncbi.nih.gov/blast/matrices/から取得することができます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f1 <- "seq2.fasta"
in_f2 <- "seq3.fasta"
param1 <- "local"
param2 <- -10
param3 <- -0.5
file <- "ftp://ftp.ncbi.nih.gov/blast/matrices/NUC.4.4"
submat <- as.matrix(read.table(file, check.names=FALSE))
library(Biostrings)
read1 <- readDNAStringSet(in_f1, format="fasta")
read2 <- readDNAStringSet(in_f2, format="fasta")
out <- pairwiseAlignment(pattern=read1,subject=read2,type=param1,
gapOpening=param2,gapExtension=param3,substitutionMatrix=submat)
out@pattern
out@subject
out@score
out_f <- "hoge.txt"
tmp <- cbind(names(read1), names(read2), out@score)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
2. multi-FASTAファイル中の「seq3 vs. seq1」のglobal alignmentの場合:
in_f <- "test2.fasta"
param1 <- "global"
param2 <- 3
param3 <- 1
param4 <- -10
param5 <- -0.5
file <- "ftp://ftp.ncbi.nih.gov/blast/matrices/NUC.4.4"
submat <- as.matrix(read.table(file, check.names=FALSE))
library(Biostrings)
reads <- readDNAStringSet(in_f, format="fasta")
out <- pairwiseAlignment(pattern=reads[param2],subject=reads[param3],type=param1,
gapOpening=param4,gapExtension=param5,substitutionMatrix=submat)
out_f <- "hoge.txt"
tmp <- cbind(names(reads[param2]), names(reads[param3]), score(out), pid(out))
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
解析 | 一般 | アラインメント | ペアワイズ | 基礎2 | Biostrings
「解析 | 一般 | アラインメント | ペアワイズ | 基礎1 | Biostrings」では、2つの配列間のアラインメントについて、その基本的な利用法とアラインメントスコアを抽出する方法について述べましたが、
他にも配列一致度など様々な情報を抽出することができます。そこで、ここでは「seq2 vs. seq3のlocal alignment結果」からどのような情報が取れるかなどを中心に手広く紹介します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f1 <- "seq2.fasta"
in_f2 <- "seq3.fasta"
param1 <- "local"
param2 <- -10
param3 <- -0.5
file <- "ftp://ftp.ncbi.nih.gov/blast/matrices/NUC.4.4"
submat <- as.matrix(read.table(file, check.names=FALSE))
library(Biostrings)
read1 <- readDNAStringSet(in_f1, format="fasta")
read2 <- readDNAStringSet(in_f2, format="fasta")
out <- pairwiseAlignment(pattern=read1,subject=read2,type=param1,
gapOpening=param2,gapExtension=param3,substitutionMatrix=submat)
out
score(out)
pattern(out)
subject(out)
nchar(out)
nmatch(out)
nmismatch(out)
nedit(out)
pid(out)
解析 | 一般 | アラインメント | ペアワイズ | 応用 | Biostrings
グローバルアラインメント(global alignment;EMBOSSのneedleに相当)や
ローカルアラインメント(local alignment; EMBOSSのwaterに相当)をやってくれます。
ここでは、3つの塩基配列(seq1, seq2, and seq3)からなるmulti-FASTA形式のファイルtest2.fastaを入力として、「それ以外 vs. 特定の配列」のペアワイズアラインメントを一気にやる方法を紹介します
ちなみにこのページでは、「配列A vs. 配列B」という表記法で、配列Bに相当するほうが"subject"で配列Aに相当するほうが"pattern"です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 特定の配列が入力multi-FASTAファイルの1番目にある場合:
in_f <- "test2.fasta"
param1 <- "local"
param2 <- 1
param3 <- -10
param4 <- -0.5
file <- "ftp://ftp.ncbi.nih.gov/blast/matrices/NUC.4.4"
submat <- as.matrix(read.table(file, check.names=FALSE))
library(Biostrings)
reads <- readDNAStringSet(in_f, format="fasta")
out <- pairwiseAlignment(pattern=reads[-param2],subject=reads[param2],type=param1,
gapOpening=param3,gapExtension=param4,substitutionMatrix=submat)
names(reads[param2])
names(reads[-param2])
length(reads[-param2])
out[1]
out[1]@score
score(out[1])
out[2]
out[2]@score
score(out[2])
score(out)
out_f <- "hoge.txt"
tmp <- NULL
for(i in 1:length(reads[-param2])){
tmp <- rbind(tmp, c(names(reads[-param2])[i], names(reads[param2]), out[i]@score))
}
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
2. 特定の配列が入力multi-FASTAファイルの3番目にある場合:
in_f <- "test2.fasta"
param1 <- "local"
param2 <- 3
param3 <- -10
param4 <- -0.5
file <- "ftp://ftp.ncbi.nih.gov/blast/matrices/NUC.4.4"
submat <- as.matrix(read.table(file, check.names=FALSE))
library(Biostrings)
reads <- readDNAStringSet(in_f, format="fasta")
out <- pairwiseAlignment(pattern=reads[-param2],subject=reads[param2],type=param1,
gapOpening=param3,gapExtension=param4,substitutionMatrix=submat)
names(reads[param2])
names(reads[-param2])
length(reads[-param2])
out[1]
out[1]@score
score(out[1])
out[2]
out[2]@score
score(out[2])
score(out)
out_f <- "hoge.txt"
tmp <- NULL
for(i in 1:length(reads[-param2])){
tmp <- rbind(tmp, c(names(reads[-param2])[i], names(reads[param2]), out[i]@score))
}
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
解析 | 一般 | アラインメント | ペアワイズ | ドットプロット | seqinr(Charif_2005)
seqinrパッケージを用いて2つの配列間のドットプロットを作成するやり方を示します。
ドットプロットとは、比較したい2つの配列をx軸y軸に並べ、同じ塩基の場所にドットをプロットするという非常にシンプルなものです。
その性質上、同一配列同士の比較の場合は対角線上にドットが打たれますが、それと異なる状況を全体的に把握したいときなどによく利用されます。
ここでは、20塩基程度までの長さのもの同士のドットプロットを描画して、乳酸菌NGS連載第13回の図2や
乳酸菌NGS連載第7回の図2と同じ結果を得るやり方を示します。
1. 同一配列("ACTCGTAGTCTATCATACGA")同士の比較の場合:
乳酸菌NGS連載第13回の図2aと同じ配列です。基本形。
seq1 <- "ACTCGTAGTCTATCATACGA"
seq2 <- "ACTCGTAGTCTATCATACGA"
library(seqinr)
dotPlot(s2c(seq1), s2c(seq2), xlab="", ylab="")
2. 同一配列("ACTCGTAGTCTATCATACGA")同士の比較の場合:
例題1と基本的に同じですが、300×300ピクセルの大きさのpngファイルとして保存するやり方です。
x軸y軸ともに、塩基配列そのものではなく数字のみの位置情報が表示されていることがわかります。
デフォルトの余白だと、本来表示させたいドットプロットの結果が小さくなってしまっています。例題3と比べるとよくわかります。
seq1 <- "ACTCGTAGTCTATCATACGA"
seq2 <- "ACTCGTAGTCTATCATACGA"
out_f <- "hoge2.png"
param_fig <- c(300, 300)
library(seqinr)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
dotPlot(s2c(seq1), s2c(seq2), xlab="", ylab="")
dev.off()
3. 同一配列("ACTCGTAGTCTATCATACGA")同士の比較の場合:
例題2と基本的に同じですが、余白を全て0にするやり方です。
例題2に比べて、ドットプロットのメインの部分がプロット全体の大部分を占めていることがわかります。
乳酸菌NGS連載第13回の図2aは、ドットプロットのみの図(hoge3.png)をベースとして、
塩基配列情報を手動で追加して作成しています。
seq1 <- "ACTCGTAGTCTATCATACGA"
seq2 <- "ACTCGTAGTCTATCATACGA"
out_f <- "hoge3.png"
param_fig <- c(300, 300)
library(seqinr)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 0, 0, 0))
dotPlot(s2c(seq1), s2c(seq2), xlab="", ylab="")
dev.off()
4. 異なる配列同士の比較の場合:
配列k("ACTCGTAGTCTATCATACGA")と配列l("ACTCGACTATCTGATTACGA")
を比較する場合です。配列kの6番目から15番目の部分配列(TAGTCTATCA)が反転(inverted)したものが配列lです。
乳酸菌NGS連載第13回の図2bと同じです。
seq1 <- "ACTCGTAGTCTATCATACGA"
seq2 <- "ACTCGACTATCTGATTACGA"
out_f <- "hoge4.png"
param_fig <- c(300, 300)
library(seqinr)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 0, 0, 0))
dotPlot(s2c(seq1), s2c(seq2), xlab="", ylab="")
dev.off()
5. 異なる配列同士の比較の場合:
例題3と基本的に同じですが、配列jは、配列k("ACTCGTAGTCTATCATACGA")の左側に"NN"を無理やり結合させたものです。
それ以外は同じですので、ドットプロットを描くと左下の原点から2塩基分だけ右にずれたようになっていることが分かります。
seq1 <- "NNACTCGTAGTCTATCATACGA"
seq2 <- "ACTCGTAGTCTATCATACGA"
out_f <- "hoge3.png"
param_fig <- c(300, 300)
library(seqinr)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 0, 0, 0))
dotPlot(s2c(seq1), s2c(seq2), xlab="", ylab="")
dev.off()
解析 | 一般 | アラインメント | マルチプル | について
マルチプルアラインメント(多重配列アラインメント)用です。
R以外:
- Clustal W (Clustal Omegaに飛ばされる):Thompson et al., Nucleic Acids Res., 1994
- T-Coffee:Notredame et al., J Mol Biol., 2000
- MAFFT:Katoh et al., Nucleic Acids Res., 2002
- MUSCLE:Edgar RC., BMC Bioinformatics, 2004
- DIALIGN-T:Subramanian et al., BMC Bioinformatics, 2005
- Kalign:Lassmann and Sonnhammer, BMC Bioinformatics, 2005
- Clustal W ver. 2.0 (Clustal Omegaに飛ばされる):Larkin et al., Bioinformatics, 2007
- DIALIGN-TX:Subramanian et al., Algorithms Mol Biol., 2008
- Kalign2:Lassmann et al., Nucleic Acids Res., 2009
- KalignP:Shu and Elofsson, Bioinformatics, 2011
- Clustal Omega:Sievers et al., Mol Syst Biol., 2011
解析 | 一般 | アラインメント | マルチプル | DECIPHER(Wright_2015)
DECIPHERパッケージを用いて多重配列アラインメント
(multiple sequence alignment; MSA)を行うやり方を示します。今のところMSA実行結果ファイルがAASTringSet形式なので距離行列情報にできていないのですが、
距離行列情報になったらIdClusters関数 --> WriteDendrogram関数と実行することで、
Newick formatという一般的な樹形図を描く際に用いるテキストファイルを作成することができます。
Newick形式のテキストファイルは、apeパッケージ中の
read.tree関数で読み込むことができます。またunroot関数を用いることでunrootedの樹形図をR上で描くことができ、write.tree関数を用いることでファイルに出力することができます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 10個の配列からなるmulti-FASTAファイル(test3.fasta)の場合:
アミノ酸配列のファイルです。
in_f <- "test3.fasta"
out_f <- "hoge1.fasta"
library(DECIPHER)
fasta <- readAAStringSet(in_f, format="fasta")
fasta
out <- AlignSeqs(fasta)
out
writeXStringSet(out, file=out_f, format="fasta", width=50)
解析 | 一般 | アラインメント | マルチプル | msa(Bodenhofer_2015)
msaパッケージを用いて多重配列アラインメント
(multiple sequence alignment; MSA)を行うやり方を示します。2016年12月29日現在、
型変換のところでunrooted treeを作成するための適切なオブジェクトにできていないため、unroot関数実行時にエラーがでます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 10個の配列からなるmulti-FASTAファイル(test3.fasta)の場合:
アミノ酸配列のファイルです。
in_f <- "test3.fasta"
out_f <- "hoge1.fasta"
param <- "ape::AAbin"
library(msa)
library(ape)
fasta <- readAAStringSet(in_f, format="fasta")
fasta
out <- msa(fasta)
out
hoge <- msaConvert(out, type=param)
hoge2 <- unroot(hoge)
writeXStringSet(out, file=out_f, format="fasta", width=50)
解析 | 一般 | Silhouette scores(シルエットスコア)
Silhouetteスコアの新たな使い道提唱論文(Zhao et al., Biol. Proc. Online, 2018)の利用法を説明します。
入力は「解析 | 発現変動 | 2群間 | 対応なし | 複製あり | TCC(Sun_2013)」などと同じく、
遺伝子発現行列データと比較したいグループラベル情報(Group1が1、Group2が2みたいなやつ)です。
出力は、Average Silhouette(AS値)というスカラー情報(1つの数値)です。
AS値の取り得る範囲は[-1, 1]で、数値が大きいほど指定したグループ間の類似度が低いことを意味し、
発現変動解析結果としてDifferentially Expressed Genes (DEGs)が沢山得られる傾向にあります。
逆に、AS値が低い(通常は-1に近い値になることはほぼ皆無で、相関係数と同じく0に近い)ほど
指定したグループ間の類似度が高いことを意味し、DEGがほとんど得られない傾向にあります。
論文中で提案している使い道としては、「発現変動解析を行ってDEGがほとんど得られなかった場合に、
サンプル間クラスタリング(SC)結果とAS値を提示して、(客観的な数値情報である)AS値が0に近い値だったのでDEGがないのは妥当だね」
みたいなdiscussionに使ってもらえればと思っています。RNA-seqカウントデータでもマイクロアレイデータでも使えます。
例題の多くは、サンプルデータ42の20,689 genes×18 samplesのリアルカウントデータ
(sample_blekhman_18.txt)を入力としています。
ヒトHomo sapiens; HS)のメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3),
チンパンジー(Pan troglodytes; PT)のメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザル(Rhesus macaque; RM)のメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
つまり、以下のような感じです。FはFemale(メス)、MはMale(オス)を表します。
ヒト(1-6列目): HSF1, HSF2, HSF3, HSM1, HSM2, and HSM3
チンパンジー(7-12列目): PTF1, PTF2, PTF3, PTM1, PTM2, and PTM3
アカゲザル(13-18列目): RMF1, RMF2, RMF3, RMM1, RMM2, and RMM3
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. HSF vs. PTFの場合:
HSF(ヒトメス)データが存在する1-3列目と、
PTF(チンパンジーメス)データが存在する 7-9
列目のデータのみ抽出してAS値を算出しています。Zhao et al., Biol. Proc. Online, 2018
のFig. 1bのHSF vs. PTFのAS値と同じ結果(AS = 0.389)が得られていることが分かります。尚、
このZhao論文中では、先に18サンプルの全データを用いてフィルタリング(低発現遺伝子の除去とユニークパターンのみにする作業)を行ったのち、
解析したい計6サンプルのサブセット抽出を行っているのでその手順に従っています。
in_f <- "sample_blekhman_18.txt"
param_subset <- c(1:3, 7:9)
param_G1 <- 3
param_G2 <- 3
library(cluster)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data <- data[,param_subset]
d <- as.dist(1 - cor(data, method="spearman"))
AS <- mean(silhouette(data.cl, d)[, "sil_width"])
AS
2. PTF vs. PTMの場合:
PTF(チンパンジーメス)データが存在する7-9列目と、
PTM(チンパンジーオス)データが存在する 10-12
列目のデータのみ抽出してAS値を算出しています。Zhao et al., Biol. Proc. Online, 2018
のFig. 1bのPTF vs. PTMのAS値と同じ結果(AS = 0.031)が得られていることが分かります。
in_f <- "sample_blekhman_18.txt"
param_subset <- c(7:9, 10:12)
param_G1 <- 3
param_G2 <- 3
library(cluster)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data <- data[,param_subset]
d <- as.dist(1 - cor(data, method="spearman"))
AS <- mean(silhouette(data.cl, d)[, "sil_width"])
AS
3. HS vs PT vs. RMの場合:
HS(ヒト)6 samples、PT(チンパンジー)6 samples、RM(アカゲザル)6 samplesの3生物種間のAS値を算出しています。
全サンプルのデータを使っているので、サブセットの抽出は行っていません。
このデータのサンプル間クラスタリング結果(Zhao et al., Biol. Proc. Online, 2018のFig. 1a)
でも3生物種明瞭に分離されていますが、高いAS値(= 0.4422661)が得られていることが分かります。
in_f <- "sample_blekhman_18.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
library(cluster)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
d <- as.dist(1 - cor(data, method="spearman"))
AS <- mean(silhouette(data.cl, d)[, "sil_width"])
AS
4. HS vs PT vs. RMの場合:
例題3と基本的に同じで、直接グループラベル情報を指し示すdata.clを作成しています。
ここではHSがG1群、PTがG2群、RMがG3群と指定したいので、どの列がどのグループに属するかをベタ書きしています。
例題3と同じAS値(= 0.4422661)が得られます。
in_f <- "sample_blekhman_18.txt"
data.cl <- c(1,1,1,1,1,1,2,2,2,2,2,2,3,3,3,3,3,3)
library(cluster)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
d <- as.dist(1 - cor(data, method="spearman"))
AS <- mean(silhouette(data.cl, d)[, "sil_width"])
AS
5. 1人目 vs 2人目 vs. 3人目の場合:
3群間比較ですが、生物種やメスオス無関係に、1人目をG1群、2人目をG2群、3人目をG3群としたdata.clを作成しています。
予想通り0に近いAS値(= -0.10115)が得られます。
in_f <- "sample_blekhman_18.txt"
data.cl <- c(1,2,3,1,2,3,1,2,3,1,2,3,1,2,3,1,2,3)
library(cluster)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
d <- as.dist(1 - cor(data, method="spearman"))
AS <- mean(silhouette(data.cl, d)[, "sil_width"])
AS
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。AS = 0.2294556になると思います。
in_f <- "data_hypodata_3vs3.txt"
param_G1 <- 3
param_G2 <- 3
library(cluster)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
d <- as.dist(1 - cor(data, method="spearman"))
AS <- mean(silhouette(data.cl, d)[, "sil_width"])
AS
解析 | 一般 | パターンマッチング
リファレンス配列(マップされる側)から文字列検索(マップする側)を行うやり方を示します。マッピングと同じです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. DHFR.fastaを入力として、"AATGCTCAGGTA"でキーワード探索を行う場合(No hitです...):
(Dihydrofolate reductase (DHFR)という塩基配列(NM_000791)のFASTA形式ファイルで、
Zinc Finger Nuclease (ZFN)認識配列(AATGCTCAGGTA)領域の探索の場合をイメージしています)
in_f <- "DHFR.fasta"
param <- "AATGCTCAGGTA"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- vmatchPattern(pattern=param, subject=fasta)
out[[1]]
2. DHFR.fastaを入力として、"CCTACTATGT"でキーワード探索を行う場合(存在することが分かっている断片配列):
in_f <- "DHFR.fasta"
param <- "CCTACTATGT"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
out <- vmatchPattern(pattern=param, subject=fasta)
out[[1]]
unlist(out)
start(unlist(out))
3. DHFR.fastaを入力として、"CCTACTATGT"でキーワード探索を行った結果をファイルに保存する場合:
in_f <- "DHFR.fasta"
out_f <- "hoge3.txt"
param <- "CCTACTATGT"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- vmatchPattern(pattern=param, subject=fasta)
out <- cbind(start(unlist(hoge)), end(unlist(hoge)))
colnames(out) <- c("start", "end")
out
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
4. multi-FASTAファイルhoge4.faを入力として、"AGG"でキーワード探索を行う場合:
in_f <- "hoge4.fa"
out_f <- "hoge4.txt"
param <- "AGG"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- vmatchPattern(pattern=param, subject=fasta)
out <- cbind(start(unlist(hoge)), end(unlist(hoge)))
colnames(out) <- c("start", "end")
rownames(out) <- names(unlist(hoge))
out
tmp <- cbind(rownames(out), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. multi-FASTAファイルhoge4.faをリファレンス配列(マップされる側)として、10リードからなるdata_seqlogo1.fastaでマッピングを行う場合:
in_f1 <- "hoge4.fa"
in_f2 <- "data_seqlogo1.fasta"
out_f <- "hoge5.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
reads <- readDNAStringSet(in_f2, format="fasta")
out <- c("in_f2", "in_f1", "start", "end")
for(i in 1:length(reads)){
hoge <- vmatchPattern(pattern=as.character(reads[i]), subject=fasta)
hoge1 <- cbind(start(unlist(hoge)), end(unlist(hoge)))
hoge2 <- names(unlist(hoge))
hoge3 <- rep(as.character(reads[i]), length(hoge2))
out <- rbind(out, cbind(hoge3, hoge2, hoge1))
}
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
6. multi-FASTAファイルhoge4.faをリファレンス配列(マップされる側)として、4リードからなるdata_reads.fastaでマッピングを行う場合:
in_f1 <- "hoge4.fa"
in_f2 <- "data_reads.fasta"
out_f <- "hoge6.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
reads <- readDNAStringSet(in_f2, format="fasta")
out <- c("in_f2", "in_f1", "start", "end")
for(i in 1:length(reads)){
hoge <- vmatchPattern(pattern=as.character(reads[i]), subject=fasta)
hoge1 <- cbind(start(unlist(hoge)), end(unlist(hoge)))
hoge2 <- names(unlist(hoge))
hoge3 <- rep(as.character(reads[i]), length(hoge2))
out <- rbind(out, cbind(hoge3, hoge2, hoge1))
}
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
7. multi-FASTAファイルhoge4.faをリファレンス配列(マップされる側)として、4リードからなるdata_reads.fastaでマッピングを行う場合(hoge1オブジェクトの作成のところの記述の仕方が若干異なる):
in_f1 <- "hoge4.fa"
in_f2 <- "data_reads.fasta"
out_f <- "hoge5.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f1, format="fasta")
reads <- readDNAStringSet(in_f2, format="fasta")
out <- c("in_f2", "in_f1", "start", "end")
for(i in 1:length(fasta)){
hoge <- matchPDict(PDict(reads), fasta[[i]])
hoge1 <- cbind(start(unlist(hoge)), end(unlist(hoge)))
hoge2 <- names(unlist(hoge))
hoge3 <- rep(names(fasta[i]), length(hoge2))
out <- rbind(out, cbind(hoge3, hoge2, hoge1))
#as.integer(coverage(hoge, 1, width(fasta[i])))
}
head(out)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=F)
解析 | 一般 | GC含量 (GC contents)
multi-FASTA形式ファイルやBSgenomeパッケージを読み込んで配列ごとのGC含量 (GC contents)を出力するやり方を示します。
出力ファイルは、「description」「CGの総数」「ACGTの総数」「配列長」「%GC含量」としています。
尚、%GC含量は「CGの総数/ACGTの総数」で計算しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "hoge4.fa"
out_f <- "hoge1.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- CG/ACGT*100
tmp <- cbind(names(fasta), CG, ACGT, width(fasta), GC_content)
colnames(tmp) <- c("description", "CG", "ACGT", "Length", "%GC_contents")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
in_f <- "h_rna.fasta"
out_f <- "hoge2.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- CG/ACGT*100
tmp <- cbind(names(fasta), CG, ACGT, width(fasta), GC_content)
colnames(tmp) <- c("description", "CG", "ACGT", "Length", "%GC_contents")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
in_f <- "test1.fasta"
out_f <- "hoge3.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- CG/ACGT*100
tmp <- cbind(names(fasta), CG, ACGT, width(fasta), GC_content)
colnames(tmp) <- c("description", "CG", "ACGT", "Length", "%GC_contents")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
4. 120MB程度のシロイヌナズナゲノムのmulti-FASTAファイル(TAIR10_chr_all.fas.gz)の場合:
in_f <- "TAIR10_chr_all.fas"
out_f <- "hoge4.txt"
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- CG/ACGT*100
tmp <- cbind(names(fasta), CG, ACGT, width(fasta), GC_content)
colnames(tmp) <- c("description", "CG", "ACGT", "Length", "%GC_contents")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
out_f <- "hoge5.txt"
param <- "BSgenome.Athaliana.TAIR.TAIR9"
library(Biostrings)
library(param, character.only=T)
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- CG/ACGT*100
tmp <- cbind(names(fasta), CG, ACGT, width(fasta), GC_content)
colnames(tmp) <- c("description", "CG", "ACGT", "Length", "%GC_contents")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
2013年12月にリリースされたGenome Reference Consortium GRCh38です。
out_f <- "hoge6.txt"
param <- "BSgenome.Hsapiens.NCBI.GRCh38"
library(Biostrings)
library(param, character.only=T)
tmp <- ls(paste("package", param, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- getSeq(genome)
names(fasta) <- seqnames(genome)
fasta
hoge <- alphabetFrequency(fasta)
#CG <- rowSums(hoge[,2:3])
#ACGT <- rowSums(hoge[,1:4])
CG <- apply(as.matrix(hoge[,2:3]), 1, sum)
ACGT <- apply(as.matrix(hoge[,1:4]), 1, sum)
GC_content <- CG/ACGT*100
tmp <- cbind(names(fasta), CG, ACGT, width(fasta), GC_content)
colnames(tmp) <- c("description", "CG", "ACGT", "Length", "%GC_contents")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
解析 | 一般 | CpGアイランドの同定 | について
脊椎動物(哺乳類)のゲノムでは、CGという2連続塩基(2-mer)の出現頻度がGC含量から予想される期待値よりも少ない
(ヒトでは1/4程度)ことがわかっています(Lander et al., Nature, 2001)。
この現象をCG抑制(CG suppression)といいます。
そんな中でもCGという2-merが相対的に多く出現する領域があり、それをCpGアイランド(CpG islands or CpG islets; CGIs)といい、
転写開始点近辺にあることが知られています(Gardiner-Garden and Frommer, J Mol Biol., 1987)。
ここでは、そのようなCGIsを同定するプログラムをリストアップします(一部そうでないものも含む)。基本ヒトゲノム用です。
R以外:
- Durbin's method(HMM-based):Durbin et al., , Cambridge University Press, 1998
- 原著論文の購読料がかかるため詳細不明:Ioshikhes and Zhang, Nat Genet., 2000
- CpGIS(window-based):Takai and Jones, Proc Natl Acad Sci U S A., 2002
- EMBOSS内のCpGplot(window-based; webtool):Olson, SA, Brief Bioinform., 2002
- CpGProD(window-based):Ponger and Mouchiroud, Bioinformatics, 2002
- CpG Island Searcher(CpGISのwebtool版だと思われるがリンク切れ):Takai and Jones, In Silico Biol., 2003
- 原著論文の購読料がかかるため詳細不明(HMM-based):Yoon and Vaidyanathan, IEEE Signal Processing Education Workshop, 2004
- CpGcluster(hybrid algorithm):Hackenberg et al., BMC Bioinformatics, 2006
- Glass's method:Glass et al., Nucleic Acids Res., 2007
- CpGIF(density-based):Sujuan et al., Bioinformation, 2008
- Irizarry's method(HMM-based):Irizarry et al., Mamm Genome, 2009
- CpG_MI(distance-/length-based):Su et al., Nucleic Acids Res., 2010
- HFS(HMM-based):Hsieh et al., Int J Biostat., 2009
- Wu's method(HMM-based):Wu et al., Biostatistics, 2010
- WordCluster(distance-/length-based):Hackenberg et al., Algorithms Mol Biol., 2011
- Elango's method(density-based):Elango and Soojin, Genetics, 2011
- CPSORL(window-based;プログラムの場所は見つけられず):Chuang et al., PLoS One, 2011
- Kakumani's method(DSP-based):Kakumani et al., Annu Int Conf IEEE Eng Med Biol Soc., 2011
- CpGPAP(hybrid algorithm; HMM-based; リンク切れ):Chuang et al., BMC Genet., 2012
- Kakumani's method(HMM-based):Kakumani et al., EURASIP J Bioinform Syst Biol., 2012
- ClusterPSO(hybrid algorithm; CpGcluster + PSO):Yang et al., PLoS One, 2016
- 3C-PSO(hybrid alrogithm):Boukelia et al., CIBB, 2016
- GaussianCpG(Gaussian model):Yu et al., BMC Genomics, 2017
- CpGTLBO:Yang et al., J Comput Biol., 2018
- Gómez-Martín's method:Gómez-Martín et al., Methods Mol Biol., 2018
解析 | 一般 | オペロン予測 | について
オペロン(operon)とは、
同じプロモータ(promoter)の制御下にある複数の遺伝子の領域のことです(Jacob et al., C R Hebd Seances Acad Sci., 1960)。
wikiのリンク先では以下のように書かれています。
「現在の状況としては、原義で言うところの1遺伝子のみからなるオペロンは遺伝子と呼び、
構造遺伝子部分はコーディング・リージョンと呼ぶのが比較的正確かつ円滑な意思疎通を産むといえるのかもしれない。」
従ってオペロン予測(operon prediction)は遺伝子予測や構造アノテーションと同じではないかという印象をもちますが、
少なくとも私が最初にみた2010年の論文(Chuang et al., Nucleic Acids Res., 2010)
では明確にoperon predictionと書かれているので、一応そのような目的に特化したプログラムをリストアップします。
基本原核生物用です。
R以外:
- hidden Markov model (HMM):Yada et al., Bioinformatics, 1999
- Ermolaeva's method:Ermolaeva et al., Nucleic Acids Res., 2001
- Zheng's method:Zheng et al., Genome Res., 2002
- Chen's method:Chen et al., Nucleic Acids Res., 2004
- Wang's method:Wang et al., Nucleic Acids Res., 2004
- OFS:Westover et al., Bioinformatics, 2005
- fuzzy guided genetic algorithm:Jacob et al., Bioinformatics, 2005
- VIMSS:Price et al., Nucleic Acids Res., 2005
- Chen's method:Chen et al., Genome Inform., 2004
- DVDA:Edwards et al., Nucleic Acids Res., 2005
- ODB:Okuda et al., Nucleic Acids Res., 2006
- Che's method:Che et al., Nucleic Acids Res., 2006
- GNM and GGM:Yan and Moult, Proteins, 2006
- Tran's method:Tran et al., Nucleic Acids Res., 2007
- Dam's method:Dam et al., Nucleic Acids Res., 2007
- SVM:Zhang et al., Comput Biol Chem., 2006
- Wang's method:Wang et al., Artif Intell Med., 2007
- UNIPOP:Li et al., J Bioinform Comput Biol., 2009
- Taboada's method:Taboada et al., Nucleic Acids Res., 2010
- Nannapaneni's method(真核生物用):Nannapaneni et al., Comput Biol Med., 2013
- Fortino's method:Fortino et al., BMC Bioinformatics, 2014
- Du's method:Du et al., Int J Data Min Bioinform., 2014
- MetaRon(メタゲノム用):Zaidi et al., BMC Genomics, 2021
- Operon Hunter:Assaf et al., Sci Rep., 2021
解析 | 一般 | Sequence logos | seqLogo
seqLogoパッケージを用いてsequence logos (Schneider and Stephens, 1990)を実行するやり方を示します。
ここでは、multi-FASTAファイルを読み込んでポジションごとの出現頻度を調べる目的で利用します。上流-35 bpにTATA boxがあることを示す目的などに利用されます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 入力ファイルがmulti-FASTA形式のファイル(test1.fasta)の場合:
in_f <- "test1.fasta"
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
seqLogo(out)
in_f <- "data_seqlogo1.fasta"
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
seqLogo(out)
得られた結果をPNG形式ファイルとして保存するやり方です。
in_f <- "data_seqlogo1.fasta"
out_f <- "hoge3.png"
param_fig <- c(600, 400)
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fasta")
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
全部で4行からなり、1行目がA, 2行目がC, 3行目がG, そして4行目がTの並びになっているという前提です。
列数は塩基配列の長さ分だけ長くなってかまいません。
in_f <- "data_seqlogo2.fasta"
library(Biostrings)
library(seqLogo)
hoge <- read.table(in_f)
out <- makePWM(hoge)
seqLogo(out)
Arabidopsisの上流500bpの配列セットです。500bpと長いため、461-500bpの範囲のみについて解析し、得られた図をファイルに保存するやり方です。
以下はダウンロードしたファイルの拡張子として、"fasta"を付加しているという前提です。
in_f <- "TAIR10_upstream_500_20101028.fasta"
out_f <- "hoge5.png"
param1 <- c(461, 500)
param2 <- 500
param_fig <- c(700, 400)
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(width(fasta) == param2)
fasta <- fasta[obj]
fasta
fasta <- subseq(fasta, param1[1], param1[2])
fasta
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
Arabidopsisの上流1000bpの配列セットです。1000bpと長いため、951-1000bpの範囲のみについて解析し、得られた図をファイルに保存するやり方です。
ファイル中の1塩基目が転写開始点からもっとも遠く離れたところで、1000塩基目が転写開始点のすぐ隣ということになります。
以下はダウンロードしたファイルの拡張子として、"fasta"を付加しているという前提です。
in_f <- "TAIR10_upstream_1000_20101104.fasta"
out_f <- "hoge6.png"
param1 <- c(951, 1000)
param2 <- 1000
param_fig <- c(800, 400)
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(width(fasta) == param2)
fasta <- fasta[obj]
fasta
fasta <- subseq(fasta, start=param1[1], end=param1[2])
fasta
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
6.と同じ結果が得られますが、転写開始点上流50bpのみを切り出して解析するというオプションにしています。
in_f <- "TAIR10_upstream_1000_20101104.fasta"
out_f <- "hoge7.png"
param1 <- 50
param2 <- 1000
param_fig <- c(800, 400)
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
obj <- as.logical(width(fasta) == param2)
fasta <- fasta[obj]
fasta
fasta <- subseq(fasta, width=param1, end=param2)
fasta
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
fasta <- readDNAStringSet(in_f, format="fasta")
table(width(fasta))
obj <- as.logical(width(fasta) != param2)
fasta <- fasta[obj]
fasta
8. FASTQ形式ファイル(SRR609266.fastq.gz)の場合:
small RNA-seqデータ(400Mb弱、11,928,428リード)です。圧縮ファイルもreadDNAStringSet関数で通常手順で読み込めます。
原著論文(Nie et al., BMC Genomics, 2013)中の記述から
GSE41841を頼りに、
SRP016842にたどりつき、
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の7を実行して得られたものが入力ファイルです。
in_f <- "SRR609266.fastq.gz"
out_f <- "hoge8.png"
param_fig <- c(800, 370)
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fastq")
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
9. FASTQ形式ファイル(hoge4.fastq.gz)の場合:
small RNA-seqデータ(280Mb弱、11,928,428リード)です。
原著論文(Nie et al., BMC Genomics, 2013)中の記述から
GSE41841を頼りに、
SRP016842にたどりつき、
前処理 | トリミング | アダプター配列除去(応用) | ShortRead(Morgan_2009)の4を実行して得られたものが入力ファイルです。
アダプター配列除去後のデータなので、リードごとに配列長が異なる場合でも読み込めるShortReadパッケージ中の
readFastq関数を用いています。
in_f <- "hoge4.fastq.gz"
out_f <- "hoge9.png"
param_fig <- c(787, 370)
library(ShortRead)
library(seqLogo)
fastq <- readFastq(in_f)
fasta <- sread(fastq)
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
前処理 | フィルタリング | 任意のリード(サブセット)を抽出
の8.を実行して得られたsmall RNA-seqデータ(100,000リード; 約16MB)です。
in_f <- "SRR609266_sub.fastq"
out_f <- "hoge10.png"
param_fig <- c(800, 370)
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fastq")
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
11. 入力ファイルがmulti-FASTA形式のファイル(cds.fna)の場合:
2019年5月13日の講義で利用した、2,311個のCDSのファイルです。見やすくすることを目的として、
最初の20塩基分のみ抽出してsequence logosを作成しています。
in_f <- "cds.fna"
out_f <- "hoge11.png"
param_length <- 20
param_fig <- c(600, 400)
library(Biostrings)
library(seqLogo)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
fasta <- subseq(fasta, start=1, end=param_length)
fasta
hoge <- consensusMatrix(fasta, as.prob=T, baseOnly=T)
out <- makePWM(hoge[1:4,])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
seqLogo(out)
dev.off()
解析 | 一般 | Sequence logos | ggseqlogo(Wagih_2017)
ggseqlogoパッケージを用いてsequence logos
(Schneider and Stephens, 1990)を実行するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 入力ファイルがmulti-FASTA形式のファイル(test1.fasta)の場合:
in_f <- "test1.fasta"
library(ggseqlogo)
解析 | 一般 | 上流配列解析 | LDSS(Yamamoto_2007)
Local Distribution of Short Sequence (LDSS)というのは、例えば手元に転写開始点上流1000塩基(upstream 1kb;下流3000塩基などでもよい)のFASTA形式の塩基配列セットがあったときに、
hexamer (6-mer)とかoctamer (8-mer)程度の短い塩基配列(short sequence)の分布を調べて、「Arabidopsis thalianaでは、"CTCTTC"というhexamerが転写開始点(Transcription Start Site; TSS)の近くにくるほどより多く出現する(参考文献1)」などの解析をしたい場合に行います。
入力データとして用いる上流 or 下流X塩基の配列セットは、配列取得 | 遺伝子の転写開始点近傍配列(上流配列など)を参考にして取得してください。
ここではArabidopsisの上流1000bpの配列セット(ファイル名:"TAIR10_upstream_1000_20101104.fasta")とラットの上流1000bpの配列セット(ファイル名:"rat_upstream_1000.fa")に対して、
pentamer (5-mer)の4^5(=1024)通りの配列一つ一つについて「上流1000bpのどこに出現したかを上流配列セット全体で出現頻度をカウントします。
この時、得られる情報はpentamerごとに「1bp目, 2bp目,...(1000 - 5 - 1)bp目の出現頻度」となるので、原著論文(参考文献1)と似た思想(全く同じというわけではありません!)でRPH, RPA, およびバックグラウンドレベルに比べて有意に局在化している短い配列(short sequences having localized distributions within upstream region)かどうかのフラグ情報を出力するやり方を示します。
- 1024通りのpentamer一つ一つについて、その「RPH, RPA, Local Distributionしているか否か」情報のみをファイル出力する場合:
- 上記に加え、全1024通りの配列のshort sequencesの実際の分布もpngファイルで出力したい場合:
尚、この方法(LDSS)はposition-sensitive typeには有効ですが、position-insensitive typeのものはアルゴリズム的に検出不可能です(参考文献6)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. pentamer(5-mer; 4^5=1024通り)でプロモータ構成モチーフ候補リストを作成したい場合:
in_f <- "TAIR10_upstream_1000_20101104.fasta"
out_f <- "hoge.txt"
param_kmer <- 5
param_fig <- c(1000, 500)
library(Biostrings)
seq <- readDNAStringSet(in_f, format="fasta")
seq
seq <- seq[width(seq) == median(width(seq))]
seq
reads <- mkAllStrings(c("A", "C", "G", "T"), param_kmer)
out <- NULL
for(i in 1:length(reads)){
tmp <- vmatchPattern(pattern=as.character(reads[i]), subject=seq)
s_posi_freq <- rle(sort(start(unlist(tmp))))
hoge <- rep(0, (width(seq[1]) - param_kmer + 1))
hoge2 <- replace(hoge, s_posi_freq$values, s_posi_freq$lengths)
out <- rbind(out, hoge2)
if(i%%10 == 0) cat(i, "/", length(reads), "finished\n")
}
rownames(out) <- reads
threshold <- apply(out,1,median) + 5*apply(out,1,mad)
obj <- apply(out,1,max) > threshold
baseline <- apply(out,1,median)
baseline[baseline < 1] <- 1
RPH <- apply(out,1,max) / baseline
RPA <- apply((out - baseline),1,sum) / apply(out,1,sum)
tmp <- cbind(rownames(out), RPH, RPA, obj)
colnames(tmp) <- c("k-mer", "Relative Peak Height (RPH)", "Relative Peak Area (RPA)", "Local Distribution")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
plot(out["TATAA",])
plot(out["GCCCA",])
2. 上記を基本としつつ組合せ数分だけ原著論文(参考文献1)Fig.1と同じような図をpngファイルで生成したい場合:
(以下をコピペすると作業ディレクトリ上に1024個のpngファイルが生成されますので注意!!)
in_f <- "TAIR10_upstream_1000_20101104.fasta"
out_f <- "hoge.txt"
param_kmer <- 5
param_fig <- c(1000, 500)
library(Biostrings)
seq <- readDNAStringSet(in_f, format="fasta")
seq
seq <- seq[width(seq) == median(width(seq))]
seq
reads <- mkAllStrings(c("A", "C", "G", "T"), param_kmer)
out <- NULL
for(i in 1:length(reads)){
tmp <- vmatchPattern(pattern=as.character(reads[i]), subject=seq)
s_posi_freq <- rle(sort(start(unlist(tmp))))
hoge <- rep(0, (width(seq[1]) - param_kmer + 1))
hoge2 <- replace(hoge, s_posi_freq$values, s_posi_freq$lengths)
out <- rbind(out, hoge2)
if(i%%10 == 0) cat(i, "/", length(reads), "finished\n")
}
rownames(out) <- reads
threshold <- apply(out,1,median) + 5*apply(out,1,mad)
obj <- apply(out,1,max) > threshold
baseline <- apply(out,1,median)
baseline[baseline < 1] <- 1
PH <- apply(out,1,max)
RPH <- PH / baseline
RPA <- apply((out - baseline),1,sum) / apply(out,1,sum)
tmp <- cbind(rownames(out), RPH, RPA, obj)
colnames(tmp) <- c("k-mer", "Relative Peak Height (RPH)", "Relative Peak Area (RPA)", "Local Distribution")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
for(i in 1:length(reads)){
out_f <- paste("result_", rownames(out)[i], ".png", sep="")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out[i,], ylim=c(0, max(c(PH[i], threshold[i]))),
ylab="Occurence", xlab="Position", type="p", pch=20, cex=0.8)
abline(h=baseline[i], col="red")
text(0, baseline[i], "baseline", col="red", adj=c(0,0))
abline(h=threshold[i], col="red")
text(0, threshold[i], "threshold(= baseline + 5*MAD)", col="red", adj=c(0,0))
dev.off()
}
in_f <- "rat_upstream_1000.fa"
out_f <- "hoge.txt"
param_kmer <- 5
param_fig <- c(1000, 500)
library(Biostrings)
seq <- readDNAStringSet(in_f, format="fasta")
seq
seq <- seq[width(seq) == median(width(seq))]
seq
reads <- mkAllStrings(c("A", "C", "G", "T"), param1)
out <- NULL
for(i in 1:length(reads)){
tmp <- vmatchPattern(pattern=as.character(reads[i]), subject=seq)
s_posi_freq <- rle(sort(start(unlist(tmp))))
hoge <- rep(0, (width(seq[1]) - param_kmer + 1))
hoge2 <- replace(hoge, s_posi_freq$values, s_posi_freq$lengths)
out <- rbind(out, hoge2)
if(i%%10 == 0) cat(i, "/", length(reads), "finished\n")
}
rownames(out) <- reads
threshold <- apply(out,1,median) + 5*apply(out,1,mad)
obj <- apply(out,1,max) > threshold
baseline <- apply(out,1,median)
baseline[baseline < 1] <- 1
PH <- apply(out,1,max)
RPH <- PH / baseline
RPA <- apply((out - baseline),1,sum) / apply(out,1,sum)
tmp <- cbind(rownames(out), RPH, RPA, obj)
colnames(tmp) <- c("k-mer", "Relative Peak Height (RPH)", "Relative Peak Area (RPA)", "Local Distribution")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
for(i in 1:length(reads)){
out_f <- paste("result_", rownames(out)[i], ".png", sep="")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out[i,], ylim=c(0, max(c(PH[i], threshold[i]))),
ylab="Occurence", xlab="Position", type="p", pch=20, cex=0.8)
abline(h=baseline[i], col="red")
text(0, baseline[i], "baseline", col="red", adj=c(0,0))
abline(h=threshold[i], col="red")
text(0, threshold[i], "threshold(= baseline + 5*MAD)", col="red", adj=c(0,0))
dev.off()
}
- LDSS:Yamamoto et al., BMC Genomics, 2007
- RAP-DB:Rice Annotation Project, Nucleic Acids Res., 2008
- TAIR:Reiser et al., Curr Protoc Bioinformatics., 2017
- PPDB:Yamamoto and Obokata, Nucleic Acids Res., 2008
- PLACE:Higo et al., Nucleic Acids Res., 1999
- RAR:Yamamoto et al., BMC Plant Biol., 2011
解析 | 一般 | 上流配列解析 | Relative Appearance Ratio(Yamamoto_2011)
手元に(Rで)マイクロアレイデータ解析のサンプルマイクロアレイデータ21の「発現変動遺伝子(DEG)の転写開始点のFASTA形式の上流配列セット(ファイル名:"seq_BAT_DEG.fa")」と「それ以外(nonDEG)の上流配列セット(ファイル名:"seq_BAT_nonDEG.fa")」
の二つのファイルがあったときに、任意のk-mer(4k通り;k=6のときは4096通り)に対して、どのk-merが発現変動と関連しているかをFisher's Exact Testを用いてp値を計算する手順を示します。
尚、ここで用いている二つのファイルはいずれも「ACGTのみからなり、配列長は1000bp」です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. pentamer(5-mer; 45=1024通り)で各k-merごとにp値とFDR値をリストアップする場合:
in_f1 <- "seq_BAT_DEG.fa"
in_f2 <- "seq_BAT_nonDEG.fa"
out_f <- "hoge.txt"
param_kmer <- 5
library(Biostrings)
seq_DEG <- readDNAStringSet(in_f1, format="fasta")
seq_DEG
seq_nonDEG <- readDNAStringSet(in_f2, format="fasta")
seq_nonDEG
reads <- mkAllStrings(c("A", "C", "G", "T"), param1)
out <- NULL
for(i in 1:length(reads)){
tmp <- vmatchPattern(pattern=as.character(reads[i]), subject=seq_DEG)
out_DEG <- length(start(unlist(tmp)))
tmp <- vmatchPattern(pattern=as.character(reads[i]), subject=seq_nonDEG)
out_nonDEG <- length(start(unlist(tmp)))
x <- c(out_nonDEG, length(seq_nonDEG), out_DEG, length(seq_DEG))
data <- matrix(x, ncol=2, byrow=T)
pvalue <- fisher.test(data)$p.value
out <- rbind(out, c(x, pvalue))
if(i%%10 == 0) cat(i, "/", length(reads), "finished\n")
}
rownames(out) <- reads
p.value <- out[,ncol(out)]
q.value <- p.adjust(p.value, method="BH")
tmp <- cbind(rownames(out), out, q.value)
colnames(tmp) <- c("k-mer", "Occurence in nonDEG", "# of nonDEG sequences", "Occurence in DEG", "# of DEG sequences", "p.value", "q.value")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
この項目周辺のカテゴライズはもはやメチャクチャですが...探そうと思ったときに意外と見つけるのが大変なものたちなので、見つけたときにリストアップしておきます。
例えば、regioneRは、
ある解析で得られたゲノム上の特定の領域群Aと領域群Bが手元にあったときに、そのオーバーラップが有意かどうかを判定するときに使えるプログラムです。
解析 | ゲノム | 領域の一致の評価 | regioneR(Gel_2016)
regioneRを用いて、
ゲノム上の特定の領域群Aと領域群Bを入力として、そのオーバーラップが有意かどうかを判定します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | 基礎 | k-mer | ゲノムサイズ推定(基礎) | qrqc
qrqcパッケージを用いてNGSデータのk-mer解析の基本的な考え方を、ゲノムサイズ推定を例に示します。
ゲノムサイズとは「トータルの塩基数」のことであり、例えばsample32_ref.fastaだと50塩基になります。
NGSデータは断片化されたリードからなり、サンプルデータ32のFASTA形式ファイル
(sample32_ngs.fasta)が仮想NGSデータになります。
これは20塩基分の断片配列(リード)を10個ランダム抽出したものだからです。
k-mer解析とは、リード長L(この場合L=20)よりも短いk個の連続塩基
(k < Lなので、L=20の場合はkは19以下になる)を生成させて、
主にその出現頻度を解析するものを指します。
L=20でk=19としたときはk-merの数は2個、
L=20でk=18としたときはk-merの数は3個、
L=20でk=17としたときはk-merの数は4個、...
一般式で書くと、k-merの数は(L-k+1)個と表現できます。1つのリードにつき(L-k+1)個分のk-mer数が生成されますので、
リード数をnとすると、n×(L-k+1)個分のk-merが生成されることになります。
もちろん、総数n×(L-k+1)個のk-merの中には全く同じ配列のものが含まれることもあるでしょう。
例えばACTGTTT, TCGGGGG, ACTGTTT, TCGGGGA, TCCGGGG, TCCGGGG, AAAACCC, TTTTAAAの場合は、k-merの総数は8個(k=7)です。
k-mer解析は、「ACTGTTTが2回, TCGGGGGが1回, TCGGGGAが1回, TCCGGGGが2回, AAAACCCが1回, TTTTAAAが1回出現」というのを調べるものです。
これが例題中の「kmer」というオブジェクトの出力結果に相当します。
k-merの種類数は6個になります。この種類数が例題中の「length(kmer)」というオブジェクトの出力結果に相当し、
ゲノムサイズの「推定値」そのものとして使えるのです。
例題では概念を説明すべく、仮想ゲノム配列の長さを制限していますが、実際にはバクテリアゲノムでも数MBと長いです。
これ(仮想ゲノム配列)を1つの長いリードLとだと考えます。そしてk-merのk値はある程度大きい数値を指定します。例えばk=50とかです。
そうすると、k-merの総数は(L-k+1)個ですが、Lが数MBという長さでk=50という状況(一般式で書くとL >> k)だと、
(L-k+1)個というk-merの総数はL個と近似できるわけです(だってL >> kなんだから)。
そして、k-merの種類数は、最大でも4^k個分だけ存在するわけですので、
そこそこ大きなk値を採用(例えばk=50とか)している限り、1つの長いリードLの塩基数と近似できます。
ここでのLは仮想ゲノム配列に相当します。
ここまでは仮想ゲノム配列をLとしてk-mer解析したときに、ゲノムサイズをk-merの種類数で近似できるという話でした。
ここからはNGSデータを入力として、どうやってゲノムサイズ推定を行うかという話になります。
k-mer解析は、NGSのリード長Lよりも短いk値の出現頻度解析を行うものです。
k=50など、そこそこ大きな値を用いてk-mer解析しますので、
実際ゲノムサイズの何倍読もうが(coverageが50Xだろうが1000Xだろうが、という意味)k-merの種類数には何の影響も与えません。
つまり、リード数をnとすると、k-merの総数はn×(L-k+1)個ですので、読めば読むほどどんどん増えていきますが、
k-merの種類数はゲノムサイズ以上には増えないのです。同じ配列を10回読もうが100回読もうが、種類が増える方向には寄与しないからです。
この項目の例題では、その感覚を身に着けるところまでを示しています。
例題中の「length(kmer)」というオブジェクトの出力結果と実際の答え(仮想ゲノム配列のサイズ)と比べてみるといいでしょう。
kが小さいときはよくわかりませんが、例題10の「10,000塩基の仮想ゲノム配列(リファレンス配列)で100Xのリードデータでk=11のk-mer解析」
くらいの規模だと実感できるでしょう。もちろん実データの場合は、シークエンスエラーが含まれます。
それはk-merの種類数を増やすネガティブな効果をもたらします。
がシークエンスエラー由来k-merも、「横軸が出現回数(k-merの種類は気にしない)、縦軸がその頻度」からなるグラフを描くことで同定することができます。
例えば、「ACTGTTTが2回, TCGGGGGが1回, TCGGGGAが1回, TCCGGGGが2回, AAAACCCが1回, TTTTAAAが1回出現」の場合は、
1回出現したものが4個(x=1, y=4)、2回出現したものが2個(x=2, y=2)といった具合でプロットできます。
例題10の出力ファイルはシークエンスエラーがない場合です。だいたい40-65回出現したk-merがほとんどを占めていることがわかります。
この分布が真のゲノム配列由来k-merの分布になります。
実際のシークエンスエラーを含むデータの場合は、この分布以外に数回しか出現しないk-merのところにもピークが出ます。
この状況は分布をみれば一目瞭然なので、ゲノムサイズ推定の場合には、例えば(実際のcoverageにも依存しますが概ね)
5回以下しか出現しなかったk-merはシークエンスエラー由来だと判断してk-merの種類数カウントから除外するなどして対応します。
以上は、谷澤靖洋氏提供情報などをもとに2016年1月6日に備忘録的にまとめたものです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
4X coverageであることが分かっているデータです。
理由は、50塩基長のリファレンス配列(仮想ゲノム配列; sample32_ref.fasta)から20塩基長のリードを10個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は20塩基以下ですので、条件を満たす19を指定しています。
in_f <- "sample32_ngs.fasta"
out_f <- "hoge1.png"
param_kmer <- 19
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""))
dev.off()
4X coverageであることが分かっているデータです。
理由は、50塩基長のリファレンス配列から20塩基長のリードを10個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は20塩基以下ですので、条件を満たす11を指定しています。
in_f <- "sample32_ngs.fasta"
out_f <- "hoge2.png"
param_kmer <- 11
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""))
dev.off()
4X coverageであることが分かっているデータです。
理由は、50塩基長のリファレンス配列から20塩基長のリードを10個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は20塩基以下ですので、条件を満たす5を指定しています。
in_f <- "sample32_ngs.fasta"
out_f <- "hoge3.png"
param_kmer <- 5
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""))
dev.off()
4X coverageであることが分かっているデータです。
理由は、1,000塩基長のリファレンス配列から20塩基長のリードを200個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は20塩基以下ですので、条件を満たす5を指定しています。
in_f <- "sample33_ngs.fasta"
out_f <- "hoge4.png"
param_kmer <- 5
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""))
dev.off()
10X coverageであることが分かっているデータです。
理由は、1,000塩基長のリファレンス配列から20塩基長のリードを500個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は20塩基以下ですので、条件を満たす5を指定しています。
in_f <- "sample34_ngs.fasta"
out_f <- "hoge5.png"
param_kmer <- 5
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""))
dev.off()
10X coverageであることが分かっているデータです。
理由は、10,000塩基長のリファレンス配列から40塩基長のリードを2,500個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は40塩基以下ですので、条件を満たす15を指定しています。
in_f <- "sample35_ngs.fasta"
out_f <- "hoge6.png"
param_kmer <- 15
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""))
dev.off()
40X coverageであることが分かっているデータです。
理由は、10,000塩基長のリファレンス配列から80塩基長のリードを5,000個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は80塩基以下ですので、条件を満たす21を指定しています。
hist関数実行時に分割数を指定すべくbreaksオプションも変更しています。
in_f <- "sample36_ngs.fasta"
out_f <- "hoge7.png"
param_kmer <- 21
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""),
breaks=max(kmer))
dev.off()
100X coverageであることが分かっているデータです。
理由は、10,000塩基長のリファレンス配列から100塩基長のリードを10,000個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は100塩基以下ですので、条件を満たす21を指定しています。
hist関数実行時に分割数を指定すべくbreaksオプションも変更しています。
in_f <- "sample37_ngs.fasta"
out_f <- "hoge8.png"
param_kmer <- 21
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""),
breaks=max(kmer))
dev.off()
100X coverageであることが分かっているデータです。
理由は、10,000塩基長のリファレンス配列から100塩基長のリードを10,000個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は100塩基以下ですので、条件を満たす31を指定しています。
hist関数実行時に分割数を指定すべくbreaksオプションも変更しています。
in_f <- "sample37_ngs.fasta"
out_f <- "hoge9.png"
param_kmer <- 31
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""),
breaks=max(kmer))
dev.off()
100X coverageであることが分かっているデータです。
理由は、10,000塩基長のリファレンス配列から100塩基長のリードを10,000個ランダム抽出したものだからです。
k-merのkの値は、リード配列長以下に設定します。この場合は100塩基以下ですので、条件を満たす11を指定しています。
hist関数実行時に分割数を指定すべくbreaksオプションも変更しています。
in_f <- "sample37_ngs.fasta"
out_f <- "hoge10.png"
param_kmer <- 11
param_fig <- c(400, 380)
library(qrqc)
hoge <- readSeqFile(in_f, type="fasta", kmer=T, k=param_kmer)
hoge
kmer <- table(hoge@kmer$kmer)
kmer
length(kmer)
table(kmer)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hist(kmer, ylab="Frequency(number of k-mers)",
xlab=paste("Number of occurences at k=", param_kmer, sep=""),
breaks=max(kmer))
dev.off()
解析 | 基礎 | 平均-分散プロット | について
よく統計解析手法の論文で出てくる図で、同一群の反復(複製)データがどれだけばらついているかを示すものです。
素人さんは何のことか分からないでしょうし、私も講習会などでは、この図よりは素人さんでも分かりやすいM-A plotで同一群のばらつきを示しますが、
こちらのほうが一般的です。
解析 | 基礎 | 平均-分散プロット(Technical replicates)
MarioniらはTechnical replicatesのデータがポアソン分布(Poisson distribution)に従うことを報告しています(Marioni et al., Genome Res., 2008)。
つまり「各遺伝子のtechnical replicatesデータの平均と分散が全体として同じ」だと言っているわけです。ここでは、横軸:平均、縦軸:分散としたプロットを描画してその傾向を眺めます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の場合:
総リード数補正を行って、群ごとにプロットしています。
in_f <- "data_marioni.txt"
out_f1 <- "hoge1_G1.txt"
out_f2 <- "hoge1_G1.png"
out_f3 <- "hoge1_G2.txt"
out_f4 <- "hoge1_G2.png"
out_f5 <- "hoge1_all.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(380, 420)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
hoge <- data[,data.cl==1]
nf <- mean(colSums(hoge))/colSums(hoge)
G1 <- sweep(hoge, 2, nf, "*")
colSums(G1)
hoge <- data[,data.cl==2]
nf <- mean(colSums(hoge))/colSums(hoge)
G2 <- sweep(hoge, 2, nf, "*")
colSums(G2)
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="blue")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G1", col="blue", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), data[,data.cl==2], hoge, MEAN, VARIANCE)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
png(out_f4, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G2", col="red", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
png(out_f5, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="blue")
par(new=T)
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", c("G1", "G2"), col=c("blue", "red"), pch=20)
dev.off()
2. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の場合:
総リード数補正を行って、2つの群をまとめてプロットしています。
in_f <- "data_marioni.txt"
out_f1 <- "hoge2.txt"
out_f2 <- "hoge2.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(380, 420)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
nf <- mean(colSums(data))/colSums(data)
normalized.count <- sweep(data, 2, nf, "*")
colSums(normalized.count)
hoge <- normalized.count
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), hoge, MEAN, VARIANCE)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="black")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G1+G2", col="black", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
3. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の場合:
総リード数補正を行って、2つの群をまとめてプロットしています。また、G2群のみのプロットも重ね書きしています。
in_f <- "data_marioni.txt"
out_f <- "hoge3.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(380, 420)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
nf <- mean(colSums(data))/colSums(data)
normalized.count <- sweep(data, 2, nf, "*")
colSums(normalized.count)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hoge <- normalized.count
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="black")
par(new=T)
MEAN <- apply(hoge[,data.cl==2], 1, mean)
VARIANCE <- apply(hoge[,data.cl==2], 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("bottomright", c("G1+G2", "G2"), col=c("black", "red"), pch=20)
dev.off()
4. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の場合:
TCCパッケージ中のiDEGES/edgeR正規化後のデータを用いて、2つの群をまとめてプロットしています。また、G2群のみのプロットも重ね書きしています。
in_f <- "data_marioni.txt"
out_f <- "hoge4.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(380, 420)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hoge <- normalized
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="black")
par(new=T)
MEAN <- apply(hoge[,data.cl==2], 1, mean)
VARIANCE <- apply(hoge[,data.cl==2], 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("bottomright", c("G1+G2", "G2"), col=c("black", "red"), pch=20)
dev.off()
5. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の場合:
TCCパッケージ中のiDEGES/edgeR正規化後のデータを用いて、2つの群をまとめてプロットしています。
iDEGES/edgeR-edgeR解析パイプライン適用後のFDR < 0.05を満たす遺伝子をマゼンタで色づけしています。
in_f <- "data_marioni.txt"
out_f <- "hoge5.png"
param_G1 <- 5
param_G2 <- 5
param_FDR <- 0.05
param_fig <- c(380, 420)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
obj <- as.logical(tcc$stat$q.value < param_FDR)
sum(tcc$stat$q.value < param_FDR)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hoge <- normalized
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+06), ylim=c(1e-02, 1e+06), col="black")
points(MEAN[obj], VARIANCE[obj], col="magenta", cex=0.1, pch=20)
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("bottomright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
6. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の場合:
例題1と基本的に同じですが、負の二項分布(Negative Binomial distribution)の式である「分散 = 平均 + Φ×平均^2」に当てはめてΦ(ふぁい、と読む)の値も計算して出力しています。
このデータは「分散 = 平均」で表されるポアソン分布に従うことが分かっているので、Φが0に近い値になることを確認しています。
in_f <- "data_marioni.txt"
out_f1 <- "hoge6_G1.txt"
out_f2 <- "hoge6_G2.txt"
param_G1 <- 5
param_G2 <- 5
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
hoge <- data[,data.cl==1]
nf <- mean(colSums(hoge))/colSums(hoge)
G1 <- sweep(hoge, 2, nf, "*")
hoge <- data[,data.cl==2]
nf <- mean(colSums(hoge))/colSums(hoge)
G2 <- sweep(hoge, 2, nf, "*")
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
PHI <- (VARIANCE - MEAN)/(MEAN^2)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE, PHI)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
PHI <- (VARIANCE - MEAN)/(MEAN^2)
tmp <- cbind(rownames(data), data[,data.cl==2], hoge, MEAN, VARIANCE, PHI)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
解析 | 基礎 | 平均-分散プロット(Biological replicates)
Biological replicatesのデータは負の二項分布(negative binomial distribution; 分散 > 平均)に従うことを検証します。
つまり、ポアソン分布(分散 = 平均)よりももっとばらつきが大きいということを言っています。
ここでは、横軸:平均、縦軸:分散としたプロットを描画してその傾向を眺めます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。2020年4月1日に負の二項分布(Negative Binomial distribution)の式である「分散 = 平均 + Φ×平均^2」に当てはめてΦ(ふぁい、と読む)の値も計算して出力するように変更しました。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge1_G1.txt"
out_f2 <- "hoge1_G1.png"
out_f3 <- "hoge1_G2.txt"
out_f4 <- "hoge1_G2.png"
out_f5 <- "hoge1_all.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(380, 420)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
hoge <- data[,data.cl==1]
nf <- mean(colSums(hoge))/colSums(hoge)
G1 <- sweep(hoge, 2, nf, "*")
colSums(G1)
hoge <- data[,data.cl==2]
nf <- mean(colSums(hoge))/colSums(hoge)
G2 <- sweep(hoge, 2, nf, "*")
colSums(G2)
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
PHI <- (VARIANCE - MEAN)/(MEAN^2)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE, PHI)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G1", col="blue", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
PHI <- (VARIANCE - MEAN)/(MEAN^2)
tmp <- cbind(rownames(data), data[,data.cl==2], hoge, MEAN, VARIANCE, PHI)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
png(out_f4, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G2", col="red", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
png(out_f5, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
par(new=T)
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", c("G1", "G2"), col=c("blue", "red"), pch=20)
dev.off()
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
non-DEGデータのみで総リード数を揃えてから計6サンプルを一緒にしてプロットしています。これが検定のイメージです。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge2.txt"
out_f2 <- "hoge2.png"
param_DEG_G1 <- 1:1800
param_DEG_G2 <- 1801:2000
param_nonDEG <- 2001:10000
param_fig <- c(380, 420)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
colSums(data[param_nonDEG,])
hoge <- data[param_nonDEG,]
nf <- mean(colSums(hoge))/colSums(hoge)
normalized <- sweep(data, 2, nf, "*")
colSums(normalized[param_nonDEG,])
hoge <- normalized
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), data, hoge, MEAN, VARIANCE)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
obj <- param_nonDEG
plot(MEAN[obj], VARIANCE[obj], log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="black")
par(new=T)
obj <- param_DEG_G1
plot(MEAN[obj], VARIANCE[obj], log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
par(new=T)
obj <- param_DEG_G2
plot(MEAN[obj], VARIANCE[obj], log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red",
xlab="MEAN", ylab="VARIANCE")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", c("DEG(G1)", "DEG(G2)", "non-DEG"), col=c("blue", "red", "black"), pch=20)
dev.off()
3. NBPSeqパッケージ中の26,222 genes×6 samplesの場合:
3 Mockサンプル vs. 3 treatedサンプルのデータです。
もう少し具体的には、バクテリアを感染させて防御応答をみたデータで、詳細はCumbie et al., PLoS One, 2011に書かれています。
out_f1 <- "hoge3_G1.txt"
out_f2 <- "hoge3_G1.png"
out_f3 <- "hoge3_G2.txt"
out_f4 <- "hoge3_G2.png"
out_f5 <- "hoge3_all.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(380, 420)
library(NBPSeq)
data(arab)
data <- arab
dim(data)
head(data)
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
hoge <- data[,data.cl==1]
nf <- mean(colSums(hoge))/colSums(hoge)
G1 <- sweep(hoge, 2, nf, "*")
colSums(G1)
hoge <- data[,data.cl==2]
nf <- mean(colSums(hoge))/colSums(hoge)
G2 <- sweep(hoge, 2, nf, "*")
colSums(G2)
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G1", col="blue", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
png(out_f4, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G2", col="red", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
png(out_f5, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
par(new=T)
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", c("G1", "G2"), col=c("blue", "red"), pch=20)
dev.off()
4. サンプルデータ8の26,221 genes×6 samplesのリアルデータ(data_arab.txt; mock 3サンプル vs. hrcc 3サンプル)の場合:
3.と基本的に同じデータですが、ファイルの読み込みから行う一般的なやり方です。
in_f <- "data_arab.txt"
out_f1 <- "hoge4_G1.txt"
out_f2 <- "hoge4_G1.png"
out_f3 <- "hoge4_G2.txt"
out_f4 <- "hoge4_G2.png"
out_f5 <- "hoge4_all.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(380, 420)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
hoge <- data[,data.cl==1]
nf <- mean(colSums(hoge))/colSums(hoge)
G1 <- sweep(hoge, 2, nf, "*")
colSums(G1)
hoge <- data[,data.cl==2]
nf <- mean(colSums(hoge))/colSums(hoge)
G2 <- sweep(hoge, 2, nf, "*")
colSums(G2)
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G1", col="blue", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
png(out_f4, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G2", col="red", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
png(out_f5, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
par(new=T)
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", c("G1", "G2"), col=c("blue", "red"), pch=20)
dev.off()
5. サンプルデータ8の26,221 genes×6 samplesのリアルデータ(data_arab.txt; mock 3サンプル vs. hrcc 3サンプル)の場合:
G1とG2群を混ぜてプロットしています。
in_f <- "data_arab.txt"
out_f <- "hoge5.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(380, 420)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
nf <- mean(colSums(data))/colSums(data)
normalized.count <- sweep(data, 2, nf, "*")
colSums(normalized.count)
hoge <- normalized.count
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="black")
par(new=T)
hoge <- normalized.count[,data.cl==1]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
par(new=T)
hoge <- normalized.count[,data.cl==2]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", c("(G1+G2)", "G1", "G2"), col=c("black", "blue", "red"), pch=20)
dev.off()
6. サンプルデータ8の26,221 genes×6 samplesのリアルデータ(data_arab.txt; mock 3サンプル vs. hrcc 3サンプル)の場合:
TCCパッケージ中のiDEGES/edgeR正規化後のデータを用いて、2つの群をまとめてプロットしています。
iDEGES/edgeR-edgeR解析パイプライン適用後のFDR < 0.05を満たす遺伝子をマゼンタで色づけしています。
in_f <- "data_arab.txt"
out_f <- "hoge6.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(380, 420)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
obj <- as.logical(tcc$stat$q.value < param_FDR)
sum(tcc$stat$q.value < param_FDR)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
hoge <- normalized
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="black")
points(MEAN[obj], VARIANCE[obj], col="magenta", cex=0.1, pch=20)
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("bottomright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
例題1と基本的に同じですが回帰分析のところを、線形回帰(分散 = a + b×平均)ではなく、
負の二項分布(Negative Binomial distribution)の式である「分散 = 平均 + Φ×平均^2」で行っています。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge7_G1.txt"
out_f2 <- "hoge7_G1.png"
out_f3 <- "hoge7_G2.txt"
out_f4 <- "hoge7_G2.png"
out_f5 <- "hoge7_all.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(380, 420)
library(MASS)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
hoge <- data[,data.cl==1]
nf <- mean(colSums(hoge))/colSums(hoge)
G1 <- sweep(hoge, 2, nf, "*")
colSums(G1)
hoge <- data[,data.cl==2]
nf <- mean(colSums(hoge))/colSums(hoge)
G2 <- sweep(hoge, 2, nf, "*")
colSums(G2)
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
PHI <- (VARIANCE - MEAN)/(MEAN^2)
tmp <- cbind(rownames(data), data[,data.cl==1], hoge, MEAN, VARIANCE, PHI)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G1", col="blue", pch=20)
obj <- as.logical(PHI > 0)
hoge <- hoge[obj,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
PHI <- (VARIANCE - MEAN)/(MEAN^2)
hoge <- as.data.frame(cbind(MEAN, VARIANCE, PHI))
out <- glm.nb(VARIANCE~MEAN+PHI, data=hoge)
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
PHI <- (VARIANCE - MEAN)/(MEAN^2)
tmp <- cbind(rownames(data), data[,data.cl==2], hoge, MEAN, VARIANCE, PHI)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
png(out_f4, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", "G2", col="red", pch=20)
hoge <- hoge[apply(hoge, 1, var) > 0,]
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
hoge <- as.data.frame(cbind(MEAN, VARIANCE))
out <- lm(VARIANCE~MEAN, data=log10(hoge))
abline(out, col="black")
out
summary(out)
dev.off()
hoge <- G1
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
png(out_f5, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1, ann=F,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="blue")
par(new=T)
hoge <- G2
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="red")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
legend("topright", c("G1", "G2"), col=c("blue", "red"), pch=20)
dev.off()
解析 | 新規転写物同定(ゲノム配列を利用)
reference-based methodsというカテゴリに含まれるものたちです。
ゲノム配列にRNA-seqデータのマッピングを行ってどこに遺伝子領域があるかなどの座標(アノテーション)情報を取得する遺伝子構造推定用です。
実質的にde novo transcriptome assemblyと目指すところは同じですが、
やはりゲノムというリファレンス配列を用いるほうがより正確であるため、ゲノム配列が利用可能な場合は利用するのが一般的です。
一般にこの種のプログラムは遺伝子構造推定だけでなく、発現量推定まで行ってくれます。
とりあえずリストアップしただけのものもあったと思いますので、間違いもいくつか含まれていると思います。
R以外:
- Scripture:Guttman et al., Nat Biotechnol., 2010
- Cufflinks:Trapnell et al., Nat Biotechnol., 2010
- rQuant: Bohnert and Ratsch, Nucleic Acids Res., 2010
- STM:Surget-Groba and Montoya-Burgos, Genome Res., 2010
- ALEXA-seq:Griffith et al., Nat Methods, 2010
- MISO: Katz et al., Nat. Methods, 2010
- MMSEQ: Turro et al., Genome Biol., 2011
- IsoEM: Nicolae et al., Algorithms Mol. Biol., 2011
- IsoformEx: Kim et al., BMC Bioinformatics, 2011
- RSEM: Li and Dewey, BMC Bioinformatics, 2011
- SLIDE: Li et al., PNAS, 2011
- IQSeq: Du et al., PLoS One, 2012
- BitSeq: Glaus et al., Bioinformatics, 2012
- ARTADE2:Kawaguchi et al., Bioinformatics, 2012
- RD:Wan et al., Biostatistics, 2012
- Plntron:Pirola et al., BMC Bioinformatics, 2012
- CEM:Li and Jiang, Bioinformatics, 2012
- iReckon:Mezlini et al., Genome Res., 2013
- TrueSight:Li et al., Nucleic Acids Res., 2013(p3)
- PASTA:Tang et al., BMC Bioinformatics, 2013
- Rockhopper(バクテリア用):McClure et al., Nucleic Acids Res., 2013
- CLASS:Song and Florea, BMC Bioinformatics, 2013
- GeneScissors:Zhang et al., Bioinformatics, 2013
- NURD:Ma et al., BMC Bioinformatics, 2013
- PSGInfer:LeGault et al., Bioinformatics, 2013
- TruHMM(バクテリア用):Li et al., BMC Genomics, 2013
- MITIE:Behr et al., Bioinformatics, 2013
- ORMAN:Dao et al., Bioinformatics, 2014
- UnSplicer:Burns et al., Nucleic Acids Res., 2014
- PennSeq:Hu et al., Nucleic Acids Res., 2014
- Parseq:Mirauta et al., Bioinformatics, 2014
- Orione(バクテリア用):Cuccuru et al., Bioinformatics, 2014
- TransComb:Liu et al., Genome Biol., 2016
- Strawberry:Liu and Dickerson, PLoS Comput Biol., 2017
- Ryūtō:Gatter and Stadler, BMC Bioinformatics, 2019
- TransBorrow:Yu et al., Genome Res., 2020
解析 | 発現量推定(トランスクリプトーム配列を利用)
新規転写物(新規isoform)の発見などが目的でなく、既知転写物の発現量を知りたいだけの場合には、やたらと時間がかかるゲノム配列へのマッピングを避けるのが一般的です。
有名なCufflinksも一応GTF形式のアノテーションファイルを与えることでゲノム全体にマップするのを避けるモードがあるらしいので、一応リストアップしています。
転写物へのマッピングの場合には、splice-aware alignerを用いたジャンクションリードのマッピングを行う必要がないので、高速にマッピング可能なbasic alignerで十分です。
但し、複数個所にマップされるリードは考慮する必要があり、確率モデルのパラメータを最尤法に基づいて推定するexpectation-maximization (EM)アルゴリズムがよく用いられます。
マッピングを行わずに、k-merを用いてalignment-freeで行う発現量推定を行うSailfishやRNA-Skimは従来法に比べて劇的に高速化がなされているようです。
間違いがいくつか含まれているとは思います。
R用:
- AllelicImbalance:Gådin et al., BMC Bioinformatics, 2015
- tximport:Soneson et al., F1000Res., 2015
- RNAontheBENCH (github上にある):Germain et al., Nucleic Acids Res., 2016
- SARTools (github上にある):Varet et al., PLoS One, 2016
- RATs:Froussios et al., F1000Res., 2019
R以外:
- Cufflinks:Trapnell et al., Nat Biotechnol., 2010
- NEUMA:Lee et al., Nucleic Acids Res., 2011
- IsoEM: Nicolae et al., Algorithms Mol. Biol., 2011
- RSEM: Li and Dewey, BMC Bioinformatics, 2011
- eXpress:Roberts and Pachter, Nat Methods, 2013
- ReXpress: Roberts et al., Bioinformatics, 2013
- TIGAR:Nariai et al., Bioinformatics, 2013
- eXpress-D:Roberts et al., BMC Bioinformatics, 2013
- PennSeq:Hu et al., Nucleic Acids Res., 2014
- Sailfish: Patro et al., Nat Biotechnol., 2014
- Quinn's pipeline(allele-specific): Quinn et al., Bioinformatics, 2014
- RNA-Skim: Zhang and Wang, Bioinformatics, 2014
- QuASAR(allele-specific): Harvey et al., Bioinformatics, 2015
- TIGAR2: Nariai et al., BMC Genomics, 2014
- SUPPA: Alamancos et al., RNA, 2015
- folded Skellam mixture model(allele-specific): Lu et al., BMC Genomics, 2015
- EMSAR: Lee et al., BMC Bioinformatics, 2015
- PGSeq: Liu et al., PLoS One, 2015
- NLDMseq: Liu et al., BMC Bioinformatics, 2015
- ASE-TIGAR(allele-specific): Nariai et al., BMC Genomics, 2016
- SplAdder: Kahles et al., Bioinformatics, 2016
- kallisto: Bray et al., Nat Biotechnol., 2016
- RapMap: Srivastava et al., Bioinformatics, 2016
- Salmon: Patro et al., Nat Methods, 2017
解析 | 融合遺伝子の同定
まずはリストアップ。
R以外:
- FusionMap:Ge et al., Bioinformatics, 2011
- TopHat-Fusion:Kim and Salzberg, Genome Biol., 2011
- FusionQ:Liu et al., BMC Bioinformatics, 2013
- ChimPipe:Rodríguez-Martín et al., BMC Genomics, 2017
- ChimeRScope(Galaxy版もあり):Li et al., Nucleic Acids Res., 2017
- MACHETE:Hsieh et al., Nucleic Acids Res., 2017
- GFusion:Zhao et al., Sci Rep., 2017
- FuSeq(non-commercial uses):Vu et al., BMC Genomics, 2018
解析 | 発現量推定(ゲノム配列を利用)
リファレンスとしてゲノム配列にマップして発現量推定を行うものたちです。だんだんカテゴリー分けが厳しくなってきましたが、とりあえず。。。
2016年10月に調べた結果をリストアップします:
解析 | 前処理 | 型変換 | について
発現データの代表的なRの格納形式として、ExpressionSet形式、SummarizedExperiment形式、そしてRangedSummarizedExperiment形式が挙げられます
(SummarizedExperimentパッケージの
htmlマニュアル)。
htmlマニュアルのリンク先でも書かれているように、
ExpressionSet形式はマイクロアレイデータ解析の頃から使われている昔ながらの形式で、
SummarizedExperimentやRangedSummarizedExperimentはRNA-seqの発現データを取り扱うようになった比較的最近(2015年頃-)の形式です。
パッケージによっては、RNA-seqカウントデータをExpressionSet形式で取り扱ったりしますが、とりあえずここで示した程度の変換例があれば事足りると思われます。
scRNA-seqデータ用の形式としては、SCESet形式やCellDataSet形式が挙げられますが、とりあえず必要最小限の情報のみ記載しています。(2019年04月03日追加)。
解析 | 前処理 | 型変換 | ExpressionSet --> SummarizedExperiment
ExpressionSetをSummarizedExperimentに変換するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
ExpressionSetオブジェクトの作成までは、「イントロ | 一般 | ExpressionSet | 1から作成 | Biobase」
の例題1と全く同じです。
in_f <- "srp001558_count_hoge6.txt"
library(Biobase)
library(SummarizedExperiment)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
eset <- ExpressionSet(assayData=as.matrix(data))
eset
rse <- as(eset, "SummarizedExperiment")
rse
解析 | 前処理 | 型変換 | ExpressionSet --> RangedSummarizedExperiment
ExpressionSetをRangedSummarizedExperimentに変換するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
ExpressionSetオブジェクトの作成までは、「イントロ | 一般 | ExpressionSet | 1から作成 | Biobase」
の例題1と全く同じです。makeSummarizedExperimentFromExpressionSet関数を利用するやり方です。
in_f <- "srp001558_count_hoge6.txt"
library(Biobase)
library(SummarizedExperiment)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
eset <- ExpressionSet(assayData=as.matrix(data))
eset
rse <- makeSummarizedExperimentFromExpressionSet(eset)
rse
例題1と基本的に同じですが、型変換に利用する関数が異なります。
in_f <- "srp001558_count_hoge6.txt"
library(Biobase)
library(SummarizedExperiment)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
eset <- ExpressionSet(assayData=as.matrix(data))
eset
rse <- as(eset, "RangedSummarizedExperiment")
rse
解析 | 前処理 | 型変換 | RangedSummarizedExperiment --> ExpressionSet
RangedSummarizedExperimentをExpressionSetに変換するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「asMethod(object) で:No assay named ‘exprs’ found, renaming counts to ‘exprs」
という警告メッセージが出ますが無視で構いません。
library(SummarizedExperiment)
example(RangedSummarizedExperiment)
rse
eset <- as(rse, "ExpressionSet")
eset
2. recountパッケージでSRP001558データのダウンロードから行う場合:
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題2の途中まで(rseオブジェクト取得まで)は基本的に同じです。
「asMethod(object) で:No assay named ‘exprs’ found, renaming counts to ‘exprs」
という警告メッセージが出ますが無視で構いません。
param_ID <- "SRP001558"
library(recount)
library(SummarizedExperiment)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
eset <- as(rse, "ExpressionSet")
eset
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題3をベースに作成しています。
「asMethod(object) で:No assay named ‘exprs’ found, renaming counts to ‘exprs」
という警告メッセージが出ますが無視で構いません。
in_f <- "rse_gene.Rdata"
library(SummarizedExperiment)
load(in_f)
rse <- rse_gene
rse
eset <- as(rse, "ExpressionSet")
eset
解析 | 前処理 | 型変換 | SCESet --> CellDataSet
scranパッケージ中のconvertTo関数を用いて、SCESetをCellDataSetに変換するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | 前処理 | フィルタリング | 低発現遺伝子 | について
直感的に当たり前ですが、低発現遺伝子は基本的に発現変動遺伝子として検出されない傾向にあります。
ここでは遺伝子発現行列(カウントデータなどの数値行列)を入力として、
発現変動解析を行う前に低発現遺伝子やサンプル間で発現変動のない遺伝子を予め除去する
プログラム群をとりあえずリストアップしています。scaterは、scRNA-seq用です。
遺伝子ごとにドロップアウト率(dropout rates)を算出する関数もあるようなので、それをフィルタリングに利用可能です。
解析 | 前処理 | フィルタリング | 低発現遺伝子 | 基礎
項目名どおりですが、発現行列データを入力として、低発現遺伝子の行をフィルタリングするやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
全6サンプル全てでカウントの総和が0以下となっている遺伝子(行)を除去する例です。
フィルタリング後に9,941 genesになっていることが分かります。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_thres <- 0
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題1と基本的に同じで、閾値が異なるだけです。
フィルタリング後に9,188 genesになっていることが分かります。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_thres <- 5
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
どちらかの群で、0カウントよりも大きいサンプル数が2つ以上ある遺伝子を抽出するやり方です。
フィルタリング後に9,315 genesになっていることが分かります。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 3
param_G2 <- 3
param_thres <- 0
param_num <- 2
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
dim(data)
out <- (data > param_thres)
obj1 <- rowSums(out[, data.cl == 1])
obj2 <- rowSums(out[, data.cl == 2])
obj <- as.logical(apply(cbind(obj1, obj2), 1, max) >= param_num)
data <- data[obj,]
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 前処理 | フィルタリング | 低発現遺伝子 | TCC(Sun_2013)
TCCパッケージ中のfilterLowCountGenes関数を用いて、
発現行列データから低発現遺伝子の行をフィルタリングするやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「総カウント数がparam_thresで指定した閾値以下となる遺伝子」を除去するやり方です。
フィルタリング後に9,188 genesになっていることが分かります。
例えば、計6サンプルで(0, 0, 0, 2, 0, 0)という発現ベクトルのgene_36は、総カウント数が2なので除去されます。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_thres <- 2
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(tcc$count)
tcc <- filterLowCountGenes(tcc, low.count=param_thres)
dim(tcc$count)
tmp <- cbind(rownames(tcc$count), tcc$count)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 前処理 | フィルタリング | 低発現遺伝子 | RangedSummarizedExperiment
ここでは、recountパッケージ(Collado-Torres et al., 2017)
経由でダウンロードしたRangedSummarizedExperiment形式の発現データ(このページではrseというオブジェクト名で統一)を出発点として、
低発現遺伝子のフィルタリングを行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. recountパッケージでSRP001558データのダウンロードから行う場合:
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題2の途中まで(rseオブジェクト取得まで)は基本的に同じです。
全サンプルの全てのカウントが0になっているものを除去するやり方です。
フィルタリング後に42,633 genesになっていることが分かります。
param_ID <- "SRP001558"
param_thres <- 0
library(recount)
library(SummarizedExperiment)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
obj <- as.logical(rowSums(data) > param_thres)
rse <- rse[obj]
rse
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題3をベースに作成しています。フィルタリング後に42,633 genesになっていることが分かります。
in_f <- "rse_gene.Rdata"
param_thres <- 0
library(recount)
library(SummarizedExperiment)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
obj <- as.logical(rowSums(data) > param_thres)
rse <- rse[obj]
rse
例題2と基本的に同じですが、最後にフィルタリング後のカウントデータをタブ区切りテキストファイルとして保存までするやり方です。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
param_thres <- 0
library(recount)
library(SummarizedExperiment)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
obj <- as.logical(rowSums(data) > param_thres)
rse <- rse[obj]
rse
data <- assays(rse)$counts
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 前処理 | フィルタリング | その他 | gene symbols情報があるもののみ | 基礎
様々なフィルタリング手段の一つとしてというよりは、同一gene symbolsを持つものをまとめる(Ensembl gene ID --> gene symbolsへのID変換)際に注意すべき点や基本的な考え方などを紹介する意味合いのほうが大きいです。
ここでは、「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」の例題6を実行して得られた、
ヒトの58,037 genes×6 samplesからなるカウントデータファイル(srp001558_count_hoge6.txt)と、
同じく例題4を実行して得られた対応表ファイル(srp001558_meta_features.txt)を読込みます。
アノテーションファイルのほうは58,037行×3列からなり、1列目がEnsembl gene ID, 2列目が長さ情報, そして3列目がgene symbols情報です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. gene symbolがNAでないもののみを抽出したい場合:
3列目にgene symbols情報がない場合はNAとなっているので、is.na関数を用いて判定しています。
但し「is.na関数はNAの要素をTRUEそれ以外をFALSEとする」のですが、実際にはNAの要素をFALSEとしたいので!is.naとしてTRUE/FALSEを逆転させています。
出力ファイル中の遺伝子数は25,450個ですね。例えば、
gene symbolsがNAとなっていた入力ファイル中の最後から2行目の"ENSG00000283698.1"が、正しくなくなっていることがわかります。
in_f1 <- "srp001558_count_hoge6.txt"
in_f2 <- "srp001558_meta_features.txt"
out_f1 <- "hoge1_count.txt"
out_f2 <- "hoge1_features.txt"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
obj <- !is.na(annot$symbol)
data <- data[obj, ]
dim(data)
tail(data)
obj <- is.element(annot$gene_id, rownames(data))
annot <- annot[obj,]
dim(annot)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
write.table(annot, out_f2, sep="\t", append=F, quote=F, row.names=F)
2. 一定の発現があり、且つgene symbolがNAでないものを抽出したい場合:
「解析 | 前処理 | フィルタリング | 低発現遺伝子 | 基礎(の例題1)」
と例題1を組み合わせるやり方です。出力ファイル中の遺伝子数は21,824個ですね。
tail(data)の結果からも全6サンプルの全てのカウントが0になっているものはなくなっているのがわかります。
in_f1 <- "srp001558_count_hoge6.txt"
in_f2 <- "srp001558_meta_features.txt"
out_f1 <- "hoge2_count.txt"
out_f2 <- "hoge2_features.txt"
param_thres <- 0
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
obj <- is.element(annot$gene_id, rownames(data))
annot <- annot[obj,]
dim(annot)
obj <- !is.na(annot$symbol)
annot <- annot[obj,]
dim(annot)
obj <- is.element(rownames(data), annot$gene_id)
data <- data[obj, ]
dim(data)
tail(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
write.table(annot, out_f2, sep="\t", append=F, quote=F, row.names=F)
3. 一定の発現があり、gene symbolがNAでなく、且つ同一IDに単一のgene symbolsが割り当てられているもののみを抽出したい場合:
例題2の結果ファイル(hoge2_features.txt)を眺めると、
同一Ensembl gene IDに対して複数個のgene symbolsが割り当てられているものがあることに気づきます(例:ENSG00000004866.19)。
これらを除去するやり方です。実際の除去手順は、hoge2_features.txtの3列目の情報(annot$symbolに相当)に対して
(を含む行番号情報を抽出し(posiに相当)、それ以外の行を抽出しています。
grep関数実行時に"\\("と書いていますが、
\\は(が特別な意味を持たないただの文字として認識させるためのおまじないのようなものです。
出力ファイルはsrp001558_count_hoge3.txtと
srp001558_features_hoge3.txtです。遺伝子数は21,329個ですね。
in_f1 <- "srp001558_count_hoge6.txt"
in_f2 <- "srp001558_meta_features.txt"
out_f1 <- "srp001558_count_hoge3.txt"
out_f2 <- "srp001558_features_hoge3.txt"
param_thres <- 0
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
obj <- is.element(annot$gene_id, rownames(data))
annot <- annot[obj,]
dim(annot)
obj <- !is.na(annot$symbol)
annot <- annot[obj,]
dim(annot)
obj <- is.element(rownames(data), annot$gene_id)
data <- data[obj, ]
dim(data)
tail(data)
posi <- grep("\\(", annot$symbol, ignore.case=T)
annot <- annot[-posi,]
dim(annot)
obj <- is.element(rownames(data), annot$gene_id)
data <- data[obj, ]
dim(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
write.table(annot, out_f2, sep="\t", append=F, quote=F, row.names=F)
解析 | 前処理 | フィルタリング | その他 | gene symbols情報があるもののみ | RangedSummarizedExperiment
ここでは、recountパッケージ(Collado-Torres et al., 2017)
経由でダウンロードしたRangedSummarizedExperiment形式の発現データ(このページではrseというオブジェクト名で統一)を出発点として、gene symbols情報がある遺伝子のフィルタリングを行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. gene symbolがNAでないもののみを抽出したい場合:
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題2の途中まで(rseオブジェクト取得まで)は基本的に同じです。
つまり、SRP001558を与えて、RangedSummarizedExperimentオブジェクトのダウンロードから行っています。
フィルタリング後に25,526 genesになっていることが分かります。
例題2と若干異なる結果になるのは、アノテーション情報のアップデートに起因すると思われます。
param_ID <- "SRP001558"
library(recount)
library(SummarizedExperiment)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
symbols <- as.character(as.vector(rowData(rse)$symbol))
obj <- !is.na(symbols)
rse <- rse[obj]
rse
2. gene symbolがNAでないもののみを抽出したい場合:
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題3をベースに作成しています。ダウンロード済みのrse_gene.Rdataを入力として読み込むやり方です。
フィルタリング後に25,450 genesになっていることが分かります。
in_f <- "rse_gene.Rdata"
library(recount)
library(SummarizedExperiment)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
symbols <- as.character(as.vector(rowData(rse)$symbol))
obj <- !is.na(symbols)
rse <- rse[obj]
rse
3. 一定の発現があり、且つgene symbolがNAでないものを抽出したい場合:
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題3をベースに作成しています。ダウンロード済みのrse_gene.Rdataを入力として読み込むやり方です。
例題1と同じく、フィルタリング後に21,880 genesになっていることが分かります。
in_f <- "rse_gene.Rdata"
param_thres <- 0
library(recount)
library(SummarizedExperiment)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
data <- assays(rse)$counts
obj <- as.logical(rowSums(data) > param_thres)
rse <- rse[obj]
rse
symbols <- as.character(as.vector(rowData(rse)$symbol))
obj <- !is.na(symbols)
rse <- rse[obj]
rse
解析 | 前処理 | ID変換 | Ensembl Gene ID中のバージョン情報を除去
バージョン情報つきのEnsembl Gene IDを含むファイルを入力として、そのバージョン情報部分のみをトリムしたファイルを出力するやり方を示します。
例えば、入力ファイル中のEnsembl Gene IDがENSG00000000003.14だったものが、出力ファイルではENSG0000000000となるような処理を行います。
これは、よく手元のEnsembl Gene IDはバージョン情報つきであるのに対して、他の情報との対応付けの際はバージョン情報を含まない状態で行わなければならない場合が多いためです。
作業手順としては、バージョン情報つきのEnsembl Gene IDの文字列をまず"."で区切り、2つに分割しています。
分割後は左側(1番目)の要素がバージョン情報のないEnsembl Gene IDに、そして右側(2番目)の要素がバージョン情報に相当します。
従って、分割後の1つ目の要素を取り出して、それを新たな行名として代入するという処理を行っています。
注意点としては、下記コード中のlength(hoge2)実行結果とlength(unique(hoge2))実行結果の違いからも推測できるように、
バージョン情報除去後にEnsembl Gene IDが同一になる行ができてしまうことが挙げられます(例えばENSG00000002586)。
例えば、例題1実行結果ファイルをrow.names=1オプションつきのread.table関数で読み込もうとしても、「重複した 'row.names' は許されません」となりますのでご注意ください。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題6実行結果ファイルと同じものです。「rownames(data) <- hoge2」のところでエラーメッセージが出ますが、出力ファイル自体は正しく作成されます。
in_f <- "srp001558_count_hoge6.txt"
out_f <- "hoge1.txt"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(rownames(data))
hoge <- strsplit(rownames(data), ".", fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", 1))
rownames(data) <- hoge2
length(hoge2)
length(unique(hoge2))
tmp <- cbind(hoge2, data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題1と基本的に同じですが、サンプル全てでカウントの総和が0以下となっている遺伝子(行)を除去してから行っています。
例題1では"ENSG00000002586"が原因で「重複した 'row.names' は許されません」となりましたが、
"ENSG00000002586.18_PAR_Y"が低発現遺伝子のフィルタリングで落とされ"ENSG00000002586.18"のみとなり、(他にもあったろうとは思いますが...)エラーが消えたことが分かります。
実際には、低発現パターンのフィルタリングはバージョン情報除去後に同一Ensembl Gene IDとなる可能性を減らすだけであり、必ずしもうまくいくわけではないのでご注意下さい。
in_f <- "srp001558_count_hoge6.txt"
out_f <- "hoge2.txt"
param_thres <- 0
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
hoge <- strsplit(rownames(data), ".", fixed=TRUE)
hoge2 <- unlist(lapply(hoge, "[[", 1))
rownames(data) <- hoge2
length(hoge2)
length(unique(hoge2))
tmp <- cbind(hoge2, data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 前処理 | ID変換 | Ensembl Gene ID --> gene symbols | 基礎
Ensembl gene IDとgene symbolsの対応表を利用して、発現行列の一番左側がEnsembl gene IDのカウントデータファイルを入力として、
同一gene symbolsのカウント情報をマージしてgene symbolsに変換した結果を返すやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「解析 | 前処理 | フィルタリング | その他 | gene symbols情報があるもののみ」
の例題3実行結果ファイルと同じものです。対応表のほうは3列目がgene symbols情報。対応表の1列目とカウントファイルの1列目がEnsembl gene IDです。
前処理済みなので比較的簡単なコードですみます。
21,329 Ensembl gene IDsが、21,163 gene symbolsになっていることがわかります。
in_f1 <- "srp001558_count_hoge3.txt"
in_f2 <- "srp001558_features_hoge3.txt"
out_f <- "hoge1.txt"
param <- "mean"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
symbols <- as.character(annot$symbol)
names(symbols) <- as.character(annot$gene_id)
unique_sym <- unique(symbols)
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題6(カウントデータ)、および例題4(対応表)実行結果ファイルと同じものです。前処理から行う例です。
58,037 Ensembl gene IDsが、25,219 gene symbolsになっていることがわかります。
低発現遺伝子のフィルタリングを行っていないので行全体でゼロカウントのもの(例:MIR3202-2)や、同一gene IDに複数のgene symbolsが割り当てられているもの
(例:67行目のc("ST7", "ST7-OT3", "MIR6132"))が含まれていることがわかります。
in_f1 <- "srp001558_count_hoge6.txt"
in_f2 <- "srp001558_meta_features.txt"
out_f <- "hoge2.txt"
param <- "mean"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
symbols <- as.character(annot$symbol)
names(symbols) <- as.character(annot$gene_id)
unique_sym <- unique(symbols)
unique_sym <- unique_sym[unique_sym != ""]
unique_sym <- unique_sym[!is.na(unique_sym)]
unique_sym <- unique_sym[!is.nan(unique_sym)]
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題2と基本的に同じで、低発現遺伝子のフィルタリングのみ追加しています。
58,037 Ensembl gene IDsが、21,649 gene symbolsになっていることがわかります。
まだ、同一gene IDに複数のgene symbolsが割り当てられているもの
(例:67行目のc("ST7", "ST7-OT3", "MIR6132"))が含まれていることがわかります。
in_f1 <- "srp001558_count_hoge6.txt"
in_f2 <- "srp001558_meta_features.txt"
out_f <- "hoge3.txt"
param <- "mean"
param_thres <- 0
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
obj <- is.element(annot$gene_id, rownames(data))
annot <- annot[obj,]
dim(annot)
symbols <- as.character(annot$symbol)
names(symbols) <- as.character(annot$gene_id)
unique_sym <- unique(symbols)
unique_sym <- unique_sym[unique_sym != ""]
unique_sym <- unique_sym[!is.na(unique_sym)]
unique_sym <- unique_sym[!is.nan(unique_sym)]
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題3と基本的に同じで、同一gene IDに複数のgene symbolsが割り当てられているものの除去を追加しています。
58,037 Ensembl gene IDsが、21,163 gene symbolsになっていることがわかります。
in_f1 <- "srp001558_count_hoge6.txt"
in_f2 <- "srp001558_meta_features.txt"
out_f <- "hoge4.txt"
param <- "mean"
param_thres <- 0
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
obj <- is.element(annot$gene_id, rownames(data))
annot <- annot[obj,]
dim(annot)
posi <- grep("\\(", annot$symbol, ignore.case=T)
annot <- annot[-posi,]
dim(annot)
obj <- is.element(rownames(data), annot$gene_id)
data <- data[obj, ]
dim(data)
symbols <- as.character(annot$symbol)
names(symbols) <- as.character(annot$gene_id)
unique_sym <- unique(symbols)
unique_sym <- unique_sym[unique_sym != ""]
unique_sym <- unique_sym[!is.na(unique_sym)]
unique_sym <- unique_sym[!is.nan(unique_sym)]
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題4と基本的に同じで、入力ファイルが異なるだけです。入力ファイルは
「カウント情報取得 | リアルデータ | SRP001540 | recount(Collado-Torres_2017)」
の例題6実行ファイルと同じです。出力ファイルはsrp001540_count_symbols.txtです
58,037 Ensembl gene IDsが、23,377 gene symbolsになっていることがわかります。
in_f1 <- "srp001540_count_yale.txt"
in_f2 <- "srp001540_meta_features.txt"
out_f <- "srp001540_count_symbols.txt"
param <- "mean"
param_thres <- 0
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
annot <- read.table(in_f2, header=TRUE, sep="\t", quote="")
dim(data)
dim(annot)
obj <- as.logical(rowSums(data) > param_thres)
data <- data[obj,]
dim(data)
obj <- is.element(annot$gene_id, rownames(data))
annot <- annot[obj,]
dim(annot)
posi <- grep("\\(", annot$symbol, ignore.case=T)
annot <- annot[-posi,]
dim(annot)
obj <- is.element(rownames(data), annot$gene_id)
data <- data[obj, ]
dim(data)
symbols <- as.character(annot$symbol)
names(symbols) <- as.character(annot$gene_id)
unique_sym <- unique(symbols)
unique_sym <- unique_sym[unique_sym != ""]
unique_sym <- unique_sym[!is.na(unique_sym)]
unique_sym <- unique_sym[!is.nan(unique_sym)]
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 前処理 | ID変換 | Ensembl Gene ID --> gene symbols | RangedSummarizedExperiment
ここでは、recountパッケージ(Collado-Torres et al., 2017)
経由でダウンロードしたRangedSummarizedExperiment形式の発現データ(このページではrseというオブジェクト名で統一)を出発点とするやり方を示します。
このrseオブジェクトはEnsembl gene IDとgene symbolsの対応情報を保持しているので、
同一gene symbolsのカウント情報をマージしてgene symbolsに変換した結果を返すやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. recountパッケージでSRP001558データのダウンロードから行う場合:
「解析 | 前処理 | フィルタリング | その他 | gene symbols情報があるもののみ | RangedSummarizedExperiment」
の例題1(gene symbolがNAでないもののフィルタリングまで)をまるごと含んでいます。
58,037 Ensembl gene IDsが、25,287 gene symbolsになっていることがわかります(2018年8月15日現在)。
param_ID <- "SRP001558"
out_f <- "hoge1.txt"
param <- "mean"
library(recount)
library(SummarizedExperiment)
download_study(param_ID, type="rse-gene", download=T)
load(file.path(param_ID, 'rse_gene.Rdata'))
rse <- rse_gene
rse
rse <- scale_counts(rse)
symbols <- as.character(as.vector(rowData(rse)$symbol))
obj <- !is.na(symbols)
rse <- rse[obj]
rse
symbols <- symbols[obj]
unique_sym <- unique(symbols)
length(unique_sym)
data <- assays(rse)$counts
data <- as.data.frame(data)
dim(data)
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「解析 | 前処理 | フィルタリング | その他 | gene symbols情報があるもののみ | RangedSummarizedExperiment」
の例題2(gene symbolがNAでないもののフィルタリングまで)をまるごと含んでいます。
58,037 Ensembl gene IDsが、25,219 gene symbolsになっていることがわかります(2018年8月15日現在)。
in_f <- "rse_gene.Rdata"
out_f <- "hoge2.txt"
param <- "mean"
library(recount)
library(SummarizedExperiment)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
symbols <- as.character(as.vector(rowData(rse)$symbol))
obj <- !is.na(symbols)
rse <- rse[obj]
rse
symbols <- symbols[obj]
unique_sym <- unique(symbols)
length(unique_sym)
data <- assays(rse)$counts
data <- as.data.frame(data)
dim(data)
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
tmp <- cbind(rownames(hoge), hoge)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題2をほぼ丸々含みますが、「カウント情報取得 | リアルデータ | SRP001558 | recount(Collado-Torres_2017)」
の例題6の最後のほうをさらに組み合わせています。つまり、
同一サンプル(technical replicates)の情報をマージした後、サンプル名を変更して出力させています。
出力は、25,219 gene symbols×6 sampleのカウントデータです。
in_f <- "rse_gene.Rdata"
out_f <- "hoge3.txt"
param <- "mean"
library(recount)
library(SummarizedExperiment)
load(in_f)
rse <- rse_gene
rse
rse <- scale_counts(rse)
symbols <- as.character(as.vector(rowData(rse)$symbol))
obj <- !is.na(symbols)
rse <- rse[obj]
rse
symbols <- symbols[obj]
unique_sym <- unique(symbols)
length(unique_sym)
data <- assays(rse)$counts
data <- as.data.frame(data)
dim(data)
hoge <- NULL
for(i in 1:length(unique_sym)){
hoge <- rbind(hoge, apply(data[which(symbols == unique_sym[i]),], 2, param, na.rm=TRUE))
}
rownames(hoge) <- unique_sym
dim(hoge)
uge <- as.data.frame(hoge)
data <- cbind(
uge$SRR032116,
uge$SRR032118 + uge$SRR032119,
uge$SRR032120 + uge$SRR032121,
uge$SRR032122 + uge$SRR032123,
uge$SRR032124 + uge$SRR032125,
uge$SRR032126 + uge$SRR032127)
colnames(data) <- c(
"HSF1", "HSF2", "HSF3",
"HSM1", "HSM2", "HSM3")
rownames(data)<- rownames(uge)
dim(data)
head(data)
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 前処理 | scRNA-seq | について
single-cell RNA-seq (scRNA-seq)のカウントデータは「疎(sparse)」です。つまり、多くの遺伝子のカウントがゼロ(0)という特徴を持ちます。
これには2つの理由があり、1つは「細胞のタイプごとに発現している遺伝子が異なるため本当に発現していない」という生物学的な理由(biological cause)によるもの。
そしてもう1つは「本当は発現しているんだけども捉えられていない」という技術的な理由(technical cause)によるものです。
特に、後者の技術的な理由でゼロカウントになることを「ドロップアウト(dropout)」と言います(Zappia et al., Genome Biol., 2017)。
データのほとんどがゼロでそれが検出されたりされなかったりするデータ(遺伝子)が多いのが特徴ですが、これがデータ解析時に悪さをするので前処理(preprocessing)が必要らしいです。
しかし、一言で前処理と言っても、実際には多様な処理が含まれます。例えば、転写物レベルのカウントデータを遺伝子レベルにつぶす(collapseする)処理や、
外部コントロールとして用いるspike-in転写物のカウント数がやたらと多い細胞のデータ(実験上のミスや細胞が死んでいるなどの理由による)の除去、
ごく少数の遺伝子のみのカウント数がそのライブラリの総カウント数の占めるようなlow-complexity librariesの存在確認、低発現遺伝子(low-abundance genes)や
ドロップアウト率の高い遺伝子(genes with high dropout rate)のフィルタリングなどが挙げられます
(McCarthy et al., Bioinformatics, 2017)。
この他にも、反復データをうまく利用して真の発現レベル(true expression levels)を推定するプログラム
(inferとかimputationとかdenoisingとかrecoverなどの単語を含むものが該当します)もこの範疇に含めてよいと思います。
"cells (細胞)"の代わりに"barcodes (バーコード)"という用語も使われます。これは、同じバーコード(well- or droplet-specific cellular barcode)に割り当てられた全てのリードが同じ細胞由来のリードに対応しないこともあるためです(Luecken and Theis, Mol Syst Biol., 2019のpage 3の右上あたり)。
例えば、あるバーコードが複数の細胞に間違ってタグ付けされたり(これを"doublet (ダブレット)"といいます)、どの細胞にも割当てられなかったりする(empty doublet/well)ことが現実にはあります。
scRNA-seqは実験プロトコルとして細胞ごとに分離してsequencingを行うのでsingle-cellなのですが、この際に完全には細胞を分離しきれないことに起因します。
doubletsは、サンプルのラベル情報がミスった状態で解析をするのと同義ですので、結構悪影響を及ぼします。
以下で「doublets同定用」と書いてあるのは、そのようなあやしいdoublets由来データを検出してくれるプログラムたちです。
ちなみにdoubletsというと2つの細胞ということになりますので、3つ以上の場合にはmultipletsになります。
それでもmultipletsということがほとんどないのは、おそらく3つ以上も混入してしまうような実験プロトコルは淘汰されているから...ですかね。
ちなみに、多くの実験プロトコルはプレート(plate)ごとあるいは液滴(droplet)ごとに「unique molecular identifier (UMI)という6-10塩基程度の長さの識別用タグ配列」仕込みます。
こうしておくことで、sequencingの際は全部を一緒にしてNGS機器にかけることができます(These libraries are pooled together for sequencing.)
(Luecken and Theis, Mol Syst Biol., 2019)。この「一緒にする(プールする)」ことをmultiplexingと言います。
scRNA-seqの生データ自体は、分離前の様々な細胞由来リード情報からなるので、一緒にされているものを分離する作業が必要になります。これをdemultiplexingといいます
他には、biological effectsの補正として細胞周期を揃えるbiological correctionもあります。scaterや
SCANPYがcell cycle scoreで線形回帰するシンプルな方法を実装しています(Luecken and Theis, Mol Syst Biol., 2019の6ページ目の右上)が、
より複雑なモデルでcell cycle補正を行うscLVMやf-scLVM(slalomパッケージの一部として実装されている)
もあります。また、細胞によって大きさが異なりますが、この大きさの違いは細胞周期に起因する転写の効果によって説明できるという主張
(McDavid, Nat Biotechnol., 2016)もあります。このcell size補正に特化したcgCorrectというプログラムもあるようです。
cgCorrectは部分的にcell cycle補正も行うことになります(Luecken and Theis, Mol Syst Biol., 2019の6ページ目の右中)。
R用:
- Seurat(通常用):Satija et al., Nat Biotechnol., 2015
- scater(通常用):McCarthy et al., Bioinformatics, 2017
- Cell Ranger(通常用; demultiplexing, 定量化まで):Zheng et al., Nat Commun., 2017
- f-scLVM(cell cycle補正用;slalomパッケージの一部として実装されている):Buettner et al., Genome Biol., 2017
- netSmooth(imputation用):Ronen and Akalin, F1000Res., 2018
- scImpute(imputation用):Li and Li, Nat Commun., 2018
- Mutual Nearest Neighbours (MNN)(data integration用;scranに組み込まれている):Haghverdi et al., Nat Biotechnol., 2018
- canonical correlation analysis (CCA)(data integration用;Seuratに組み込まれている):Butler et al., Nat Biotechnol., 2018
- DrImpute(imputation用):Gong et al., BMC Bioinformatics, 2018
- SAVER(imputation用):Huang et al., Nat Methods, 2018
- MAGIC(imputation用):van Dijk et al., Cell, 2018
- scPipe(通常用):Tian et al., PLoS Comput Biol., 2018
- LSImpute(imputation用; RのShiny app):Moussa and Măndoiu, J Comput Biol., 2019
- DoubletFinder(doublets同定用):McGinnis et al., Cell Syst., 2019
- scruff(通常用; CEL-Seq or CEL-Seq2データ専用?!):Wang et al., BMC Bioinformatics, 2019
- EnImpute(imputation用):Zhang et al., Bioinformatics, 2019
- LIGER(data integration用):Welch et al., Cell, 2019
- RESCUE(imputation用):Tracy et al., BMC Bioinformatics, 2019
- scds(doublets同定用):Bais and Kostka, Bioinformatics, 2019
- DoubletDecon(doublets同定用):DePasquale et al., Cell Rep., 2019
- Harmony(data integration用):Korsunsky et al., Nat Methods, 2019
- scRMD(imputation用):Chen et al., Bioinformatics, 2020
- RISC(data integration用;CRANで提供予定だそうです):Liu et al., bioRxiv, 2018
- SoupX(droplet-based用;ambient RNA contamination除去用):Young and Behjati, bioRxiv, 2020
R以外:
- scLVM(cell cycle補正用):Buettner et al., Nat Biotechnol., 2015
- inDrops(通常用; demultiplexing, 定量化まで):Klein et al., Cell, 2015
- cgCorrect(cell size補正用;プログラムは非公開?!):Blasi et al., Phys Biol., 2017
- SCANPY(通常用):Wolf et al., Genome Biol., 2018
- zUMIs(通常用; demultiplexing, 定量化まで):Parekh et al., Gigascience, 2018
- BISCUIT(imputation用;プログラムは非公開?!):Prabhakaran et al., JMLR Workshop Conf Proc., 2016やAzizi et al., Genom Comput Biol., 2017
- MAGIC(imputation用):van Dijk et al., Cell, 2018
- SEQC(通常用; demultiplexing, 定量化まで):Azizi et al., Cell, 2018
- AutoImpute(imputation用):Talwar et al., Sci Rep., 2018
- scVI(通常用):Lopez et al., Nat Methods, 2018
- VASC(imputation用):Wang and Gu, Genomics Proteomics Bioinformatics, 2018
- DCA(imputation用):Eraslan et al., Nat Commun., 2019
- McImpute(imputation用):Mongia et al., Front Genet., 2019
- Scrublet(doublets同定用):Wolock et al., Cell Syst., 2019
- scanorama(data integration用):Hie et al., Nat Biotechnol., 2019
- scGen(data integration用):Lotfollahi et al., Nat Methods, 2019
- scHinter(imputation用):Ye et al., Bioinformatics, 2019
- BBKNN(data integration用):Polański et al., Bioinformatics, 2019
- SAVER-X(imputation用):Wang et al., Nat Methods, 2019
- DeepImpute(imputation用):Arisdakessian et al., Genome Biol., 2019
- kNN-smoothing(imputation用):Wagner et al., bioRxiv, 2017
- scScope(imputation用):Deng et al., bioRxiv, 2018
解析 | クラスタリング | RNA-seq | について
だいぶ充実してきましたね。2019年4月4日にscRNA-seq用を別項目に移動させ、scRNA-seqとの対比で、(bulk) RNA-seq用のみであることを明記した項目名に変更しました。
R用:
- MBCluster.Seq(遺伝子クラスタリング):Si et al., Bioinformatics, 2014
- dendextend:Galili T, Bioinformatics, 2015
- ctsGE:Sharabi-Schwager, Bioinformatics, 2017
- NB.MClust(サンプルクラスタリング):Li et al., BMC Bioinformatics, 2018
- MPLNClust(遺伝子クラスタリングインストールが厄介そう):Silva et al., BMC Bioinformatics, 2019
R以外:
- bi-Poisson model:Wang et al., Brief Bioinform, 2014
- multi-Poisson dynamic mixture model:Ye et al., Brief Bioinform, 2015
- a two-stage EM algorithm:Jiang et al., BMC Genomics, 2014
- EPIG-Seq:Li and Bushel, BMC Genomics, 2016
- Clusternomics(Rかもだが不親切):Gabasova et al., PLoS Comput Biol., 2017
- Shrinkage Clustering:Hu et al., BMC Bioinformatics, 2018
- revamp(Lemon-Tree projectの一部?!;多分遺伝子クラスタリング):Erola et al., Bioinformatics, 2020
解析 | クラスタリング | RNA-seq | サンプル間 | hclust
RNA-seqカウントデータのクラスタリング結果は、特にゼロカウント(0カウント; zero count)を多く含む場合に(もちろん距離の定義の仕方によっても変わってきますが)
低発現データのフィルタリングの閾値次第で結果が変わる傾向にあります。
ここでは、上記閾値問題に悩まされることなく頑健なサンプル間クラスタリングを行うやり方を示します。
内部的に行っていることは、1)全サンプルで0カウントとなる行(遺伝子)をフィルタリングした後、unique関数を用いて同一発現パターンのものを1つのパターンとしてまとめ、
2)「1 - Spearman順位相関係数」でサンプル間距離を定義、3)Average-linkage clusteringの実行、です。
順位相関係数を用いてサンプルベクトル間の類似度として定義するので、サンプル間正規化の問題に悩まされません。
また、低発現遺伝子にありがちな同一発現パターンの遺伝子をまとめることで、(変動しやすい)同順位となる大量の遺伝子が集約されるため、
結果的に「総カウント数がx個以下のものをフィルタリング...」という閾値問題をクリアしたことになります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data.dist <- as.dist(1 - cor(data, method="spearman"))
out <- hclust(data.dist, method="average")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。non-DEGデータのみでクラスタリングを行っています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_nonDEG <- 2001:10000
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- data[param_nonDEG,]
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data.dist <- as.dist(1 - cor(data, method="spearman"))
out <- hclust(data.dist, method="average")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
Study列が"bodymap"データのクラスタリングを行います。"Count table"列のカウントデータファイル(bodymap_count_table.txt)
のみではサンプル名がIDとして与えられているため、"Phenotype table"列のサンプル名情報ファイル(bodymap_phenodata.txt)
も利用してbrain, liver, heartなどに変換した上でクラスタリング結果を表示させています。
in_f1 <- "bodymap_count_table.txt"
in_f2 <- "bodymap_phenodata.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
phenotype <- read.table(in_f2, header=TRUE, row.names=1, sep=" ", quote="")
colnames(data) <- phenotype$tissue.type
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data.dist <- as.dist(1 - cor(data, method="spearman"))
out <- hclust(data.dist, method="average")
png(out_f, pointsize=15, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
4. サンプルデータ8の26,221 genes×6 samplesのリアルデータ(data_arab.txt; mock 3サンプル vs. hrcc 3サンプル)の場合:
in_f <- "data_arab.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data.dist <- as.dist(1 - cor(data, method="spearman"))
out <- hclust(data.dist, method="average")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
5. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
Neyret-Kahn et al., Genome Res., 2013の2群間比較用ヒトRNA-seqカウントデータです。
パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)から得られます。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data.dist <- as.dist(1 - cor(data, method="spearman"))
out <- hclust(data.dist, method="average")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
解析 | クラスタリング | RNA-seq | サンプル間 | TCC(Sun_2013)
TCCパッケージを用いてサンプル間クラスタリングを行うやり方を示します。
clusterSample関数を利用した頑健なクラスタリング結果を返します。多群間比較用の推奨ガイドライン提唱論文
(Tang et al., BMC Bioinformatics, 2015)中でも
この関数を用いています(2015/11/05追加)。xlsx形式ファイルを入力とするやり方も追加しました(2015/11/15)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Neyret-Kahn et al., Genome Res., 2013の2群間比較用(3 proliferative samples vs. 3 Ras samples)ヒトRNA-seqカウントデータです。
パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)から得られます。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
1.との違いはplot関数実行時のオプションの部分のみです。sub=""で、下のほうの「hclust(*, "average")」という文字を消しています。
xlab=""で、下のほうの「d」という文字を消しています。cex.lab=1.5で、表示されているy軸名「Height」の文字の大きさを通常の0.6倍にし、
入力ファイル中のサンプル名の文字の大きさを通常の1.3倍にしています。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, sub="", xlab="", cex.lab=0.6, cex=1.3)
dev.off()
2.との違いはplot関数実行時のオプションの部分のみです。
ylab="height kamo"で、y軸名を通常の1.2倍の「height kamo」にしています。
main=""で、図の上のほうのメインタイトル名が表示されないようにしてます。
par(mar=c(0, 4, 1, 0))で、図の上下の余白を狭くして、図の左側のみ4行分、上を1行分空け、下と右を0行分だけ開けるように指定しています。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="height kamo")
dev.off()
このデータは2つの大きなクラスターに分かれていることが分かります。
ここでは指定したクラスター数に分けた場合に、含まれるメンバーなどを得るやり方を示します。
in_f <- "srp017142_count_bowtie.txt"
out_f1 <- "hoge4.txt"
out_f2 <- "hoge4.png"
param_fig <- c(500, 400)
param_clust_num <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
hoge <- cutree(out, k=param_clust_num)
hoge
tmp <- cbind(colnames(data), hoge)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F, col.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="Height")
dev.off()
Neyret-Kahn et al., Genome Res., 2013のgene-levelの2群間比較用(3 proliferative samples vs. 3 Ras samples)ヒトRNA-seqカウントデータです。
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)から得られます。
in_f <- "hoge9_count_gene.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="Height")
dev.off()
Neyret-Kahn et al., Genome Res., 2013のexon-levelの2群間比較用(3 proliferative samples vs. 3 Ras samples)ヒトRNA-seqカウントデータです。
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)から得られます。
in_f <- "hoge9_count_exon.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="Height")
dev.off()
Blekhman et al., Genome Res., 2010の
20,689 genes×36 samplesのカウントデータです。
in_f <- "sample_blekhman_36.txt"
out_f <- "hoge7.png"
param_fig <- c(700, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="Height")
dev.off()
Blekhman et al., Genome Res., 2010の
20,689 genes×18 samplesのカウントデータです。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge8.png"
param_fig <- c(700, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="Height")
dev.off()
8.と基本的に同じですが、図の大きさなどを変更しています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge9.png"
param_fig <- c(340, 200)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 0, 0, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", col.main="red")
dev.off()
9.と基本的に同じですが、図の大きさを変更しています。
また、余白のパラメータparam_marを追加しています。左側の軸が見えるようにしています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge10.png"
param_fig <- c(403, 200)
param_mar <- c(0, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", col.main="red")
dev.off()
Additional file 1として提供されているxlsxファイルです。
2群間比較用(8 samples vs. 8 samples)データです。余分な情報を最初の1行分に含みます。
このため、最初の2行目から読み込むやり方です。
in_f <- "s12864-015-1767-y-s1.xlsx"
out_f <- "hoge11.png"
param_skip <- 2
param_fig <- c(403, 200)
param_mar <- c(0, 4, 0, 0)
library(openxlsx)
library(TCC)
data <- read.xlsx(in_f, startRow=param_skip, colNames=T, rowNames=T)
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", col.main="red")
dev.off()
解析 | クラスタリング | RNA-seq | 遺伝子間(基礎) | MBCluster.Seq(Si_2014)
MBCluster.Seqパッケージを用いたやり方を示します。
k-means++(Arthur and Vassilvitskii, 2007)
と似た方法でクラスター中心を決める方法を内部的に利用しているらしいです。
param_clust_numで指定するクラスター数は最初は気持ち多めにしておいても、クラスタリング実行中に重複のないパターン(non-redundant expression patterns)にある程度はしてくれます。
例えば、これを利用した最近の論文(Casero et al., Nat Immunol., 2015)では、
おそらくparam_clust_numで指定するクラスター数を50として実行し、最終的に20 clustersを得たようです。
モデルはPoissonとnegative binomial (NB) modelから選択できるらしいですが、基本は後者の利用だと思います。
遺伝子間クラスタリングなのでデータの正規化が重要になってきます。
RNASeq.Data関数のところで、Normalizer=NULLにするとlog(Q2)でデータを揃えるらしいです。
この関数は、遺伝子(行)ごとの正規化後の平均発現量、正規化係数、logFC、NB.Dispersionなどクラスタリングに必要な結果を返します。
ここでは、例題7まではNormalizer=NULLに固定し、それ以外の入力ファイルや指定するクラスター数の例を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
このデータは事実上3つのクラスター(G1で高発現のDEGクラスター、G2で高発現のDEGクラスター、non-DEGクラスター)しか存在しない例ですが、
とりあえずクラスター数を4にしています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.png"
param_fig <- c(800, 500)
param_clust_num <- 4
param_G1 <- 3
param_G2 <- 3
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
dim(data)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。このデータは事実上4つのクラスター
(G1で高発現のDEGクラスター、G2で高発現のDEGクラスター、G3で高発現のDEGクラスター、non-DEGクラスター)しか存在しない例ですが、
とりあえずクラスター数を6にしています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge2.png"
param_fig <- c(800, 500)
param_clust_num <- 6
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
dim(data)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。とりあえずクラスター数を5にしています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge3.png"
param_fig <- c(800, 500)
param_clust_num <- 5
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
dim(data)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
3.と基本的に同じですが、遺伝子ごとにparam_clust_numで指定したクラスターのどこに割り振られたかの情報を、
クラスターごとにソートして保存しています。とりあえずクラスター数を10にしています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge4.png"
out_f2 <- "hoge4.txt"
param_fig <- c(800, 500)
param_clust_num <- 10
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
dim(data)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
table(cls$cluster)
png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
tmp <- cbind(rownames(data), data, cls$cluster)
tmp <- tmp[order(cls$cluster),]
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
例題2のシミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。このデータは事実上4つのクラスター
(G1で高発現のDEGクラスター、G2で高発現のDEGクラスター、G3で高発現のDEGクラスター、non-DEGクラスター)しか存在しない例です。
クラスター数を4にして、どのクラスターに割り振られたかが分かるようにしています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge5.txt"
param_clust_num <- 4
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
dim(data)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
tmp <- cbind(rownames(data), data, cls$cluster)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題5と基本的に同じですが、クラスター番号情報だけではなく、既知の発現パターン
("DEG_G1", "DEG_G2", "DEG_G3", "nonDEG")に割り当てた結果を一番右側の列に追加しています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge6.txt"
param_clust_num <- 4
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
dim(data)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
DEG_G1_posi <- 1:2100
DEG_G2_posi <- 2101:2700
DEG_G3_posi <- 2701:3000
nonDEG_posi <- 3001:10000
conversion <- 1:param_clust_num
names(conversion) <- 1:param_clust_num
pattern <- cls$cluster
hoge <- table(cls$cluster[nonDEG_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[1]
conversion[cls_num] <- param_narabi[1]
hoge <- table(cls$cluster[DEG_G1_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[2]
conversion[cls_num] <- param_narabi[2]
hoge <- table(cls$cluster[DEG_G2_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[3]
conversion[cls_num] <- param_narabi[3]
hoge <- table(cls$cluster[DEG_G3_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[4]
conversion[cls_num] <- param_narabi[4]
conversion
table(cls$cluster[nonDEG_posi])
table(cls$cluster[DEG_G1_posi])
table(cls$cluster[DEG_G2_posi])
table(cls$cluster[DEG_G3_posi])
tmp <- cbind(rownames(data), data, cls$cluster, pattern)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
7. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題6と同じ結果になります。サンプルデータ15の最初の部分を丸々含みます。
混同行列(confusion matrix)情報のファイルも出力しています。
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7_confusion.txt"
param_clust_num <- 4
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
library(TCC)
library(MBCluster.Seq)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
head(hoge$Normalizer, n=1)
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
conversion <- 1:param_clust_num
names(conversion) <- 1:param_clust_num
pattern <- cls$cluster
hoge <- table(cls$cluster[nonDEG_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[1]
conversion[cls_num] <- param_narabi[1]
hoge <- table(cls$cluster[DEG_G1_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[2]
conversion[cls_num] <- param_narabi[2]
hoge <- table(cls$cluster[DEG_G2_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[3]
conversion[cls_num] <- param_narabi[3]
hoge <- table(cls$cluster[DEG_G3_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[4]
conversion[cls_num] <- param_narabi[4]
confusion <- matrix(0, nrow=param_clust_num, ncol=param_clust_num)
for(i in 1:param_clust_num){
posi <- which(conversion == param_narabi[i])
confusion[i, 1] <- sum(cls$cluster[nonDEG_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 2] <- sum(cls$cluster[DEG_G1_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 3] <- sum(cls$cluster[DEG_G2_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 4] <- sum(cls$cluster[DEG_G3_posi] == posi)
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, cls$cluster, pattern)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
8. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題7を基本として、TMM正規化によって得られたサイズファクターをRNASeq.Data関数での読み込み時に与えています。
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8_confusion.txt"
param_clust_num <- 4
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
library(TCC)
library(MBCluster.Seq)
library(edgeR)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
#tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
#norm.factors <- tcc$norm.factors
d <- DGEList(counts=data,group=data.cl)
d <- calcNormFactors(d)
norm.factors <- d$samples$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
head(hoge$Normalizer, n=1)
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
conversion <- 1:param_clust_num
names(conversion) <- 1:param_clust_num
pattern <- cls$cluster
hoge <- table(cls$cluster[nonDEG_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[1]
conversion[cls_num] <- param_narabi[1]
hoge <- table(cls$cluster[DEG_G1_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[2]
conversion[cls_num] <- param_narabi[2]
hoge <- table(cls$cluster[DEG_G2_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[3]
conversion[cls_num] <- param_narabi[3]
hoge <- table(cls$cluster[DEG_G3_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[4]
conversion[cls_num] <- param_narabi[4]
confusion <- matrix(0, nrow=param_clust_num, ncol=param_clust_num)
for(i in 1:param_clust_num){
posi <- which(conversion == param_narabi[i])
confusion[i, 1] <- sum(cls$cluster[nonDEG_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 2] <- sum(cls$cluster[DEG_G1_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 3] <- sum(cls$cluster[DEG_G2_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 4] <- sum(cls$cluster[DEG_G3_posi] == posi)
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, cls$cluster, pattern)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
9. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題8と基本的に同じ結果になりますが、TMM正規化をedgeRパッケージ中の関数ではなく、
TCCで行っています。
out_f1 <- "hoge9.txt"
out_f2 <- "hoge9_confusion.txt"
param_clust_num <- 4
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
library(TCC)
library(MBCluster.Seq)
library(edgeR)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
norm.factors <- tcc$norm.factors
#d <- DGEList(counts=data,group=data.cl)
#d <- calcNormFactors(d)
#norm.factors <- d$samples$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
head(hoge$Normalizer, n=1)
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
conversion <- 1:param_clust_num
names(conversion) <- 1:param_clust_num
pattern <- cls$cluster
hoge <- table(cls$cluster[nonDEG_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[1]
conversion[cls_num] <- param_narabi[1]
hoge <- table(cls$cluster[DEG_G1_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[2]
conversion[cls_num] <- param_narabi[2]
hoge <- table(cls$cluster[DEG_G2_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[3]
conversion[cls_num] <- param_narabi[3]
hoge <- table(cls$cluster[DEG_G3_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[4]
conversion[cls_num] <- param_narabi[4]
confusion <- matrix(0, nrow=param_clust_num, ncol=param_clust_num)
for(i in 1:param_clust_num){
posi <- which(conversion == param_narabi[i])
confusion[i, 1] <- sum(cls$cluster[nonDEG_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 2] <- sum(cls$cluster[DEG_G1_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 3] <- sum(cls$cluster[DEG_G2_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 4] <- sum(cls$cluster[DEG_G3_posi] == posi)
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, cls$cluster, pattern)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
10. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題9はparam_narabiで示す発現パターン分類を(シミュレーションデータなので)答えが分かっているものから逆算して割り当てています。
それに対して、ここではクラスタリング結果のclsオブジェクト中に含まれる(param_clust_num)個の代表発現パターン
(これはcls$centersから取得可能)から、どのクラスター番号のものがどのパターンに相当するかを割り当てるやり方です。
out_f1 <- "hoge10.txt"
out_f2 <- "hoge10_confusion.txt"
param_clust_num <- 4
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
library(TCC)
library(MBCluster.Seq)
library(edgeR)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
norm.factors <- tcc$norm.factors
#d <- DGEList(counts=data,group=data.cl)
#d <- calcNormFactors(d)
#norm.factors <- d$samples$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
head(hoge$Normalizer, n=1)
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
cls$centers
conversion <- rep(0, param_clust_num)
conversion <- paste("DEG_G", max.col(cls$centers), sep="")
tmp <- apply(cls$centers, 1, max) - apply(cls$centers, 1, min)
obj <- (tmp == min(tmp))
conversion[obj] <- "nonDEG"
conversion
pattern <- cls$cluster
for(i in 1:param_clust_num){
pattern[pattern == i] <- conversion[i]
}
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
confusion <- matrix(0, nrow=param_clust_num, ncol=param_clust_num)
for(i in 1:param_clust_num){
posi <- which(conversion == param_narabi[i])
confusion[i, 1] <- sum(cls$cluster[nonDEG_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 2] <- sum(cls$cluster[DEG_G1_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 3] <- sum(cls$cluster[DEG_G2_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 4] <- sum(cls$cluster[DEG_G3_posi] == posi)
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, cls$cluster, pattern)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
11. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題10を基本として、nonDEGパターンに割り振られる確率でAUCを計算しています。
AUC値は0.9446149ですね。
out_f1 <- "hoge11.txt"
out_f2 <- "hoge11_confusion.txt"
param_clust_num <- 4
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
library(TCC)
library(MBCluster.Seq)
library(edgeR)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
norm.factors <- tcc$norm.factors
#d <- DGEList(counts=data,group=data.cl)
#d <- calcNormFactors(d)
#norm.factors <- d$samples$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
head(hoge$Normalizer, n=1)
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
cls$centers
conversion <- rep(0, param_clust_num)
conversion <- paste("DEG_G", max.col(cls$centers), sep="")
tmp <- apply(cls$centers, 1, max) - apply(cls$centers, 1, min)
obj <- (tmp == min(tmp))
conversion[obj] <- "nonDEG"
conversion
ranking <- rank(cls$probability[,obj])
pattern <- cls$cluster
for(i in 1:param_clust_num){
pattern[pattern == i] <- conversion[i]
}
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
confusion <- matrix(0, nrow=param_clust_num, ncol=param_clust_num)
for(i in 1:param_clust_num){
posi <- which(conversion == param_narabi[i])
confusion[i, 1] <- sum(cls$cluster[nonDEG_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 2] <- sum(cls$cluster[DEG_G1_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 3] <- sum(cls$cluster[DEG_G2_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 4] <- sum(cls$cluster[DEG_G3_posi] == posi)
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, cls$cluster, pattern)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | クラスタリング | RNA-seq | 遺伝子間(応用) | TCC正規化(Sun_2013)+MBCluster.Seq(Si_2014)
MBCluster.Seqパッケージを用いたやり方を示します。
遺伝子間クラスタリングなのでデータの正規化が重要になってきます。ここでは、
TCCを実行して得られたTCC正規化係数(iDEGES/edgeR正規化を実行して得られたサンプルごとに与える係数)
を入力として与えるやり方を示します。正確には、
DESeqパッケージと同じようにsize.factorsに変換してから与えています。
理由は、TCCや
edgeRパッケージ中で取り扱っている「正規化係数(normalization factors)」は、
「総リード数(library sizes)」を補正するための「係数」という位置づけだからです(正規化係数を掛けたあとのライブラリサイズをeffective library sizesともいう)。
その一方でMBCluster.Seq
中で述べている正規化は、DESeqやDESeq2で使われている。
つまり、TCC正規化係数*総リード数 = effective library sizes、そしてeffective library sizesをさらに正規化したものがsize factorsという関係になります。
2021年5月に精査した結果、RNASeq.Data関数のNormalizerオプションで与える情報は「size factorsそのもの」や「そのlog」ではなく「log2」が正解のようですのでそのように修正しましたm(_ _)m
尚、この場合は結果的にeffective library sizesそのものから算出しても同じなのでそのようにしています。(2021年5月28日追加)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
このデータは事実上3つのクラスター(G1で高発現のDEGクラスター、G2で高発現のDEGクラスター、non-DEGクラスター)しか存在しない例ですが、
とりあえずクラスター数を4にしています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.png"
param_fig <- c(800, 500)
param_clust_num <- 4
param_G1 <- 3
param_G2 <- 3
library(MBCluster.Seq)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
1.と基本的に同じですが、遺伝子ごとにparam_clust_numで指定したクラスターのどこに割り振られたかの情報もテキストファイルに保存するやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge2.png"
out_f2 <- "hoge2.txt"
param_fig <- c(800, 500)
param_clust_num <- 4
param_G1 <- 3
param_G2 <- 3
library(MBCluster.Seq)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
table(cls$cluster)
png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(data), normalized, cls$cluster)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
2.と基本的に同じですが、遺伝子ごとにparam_clust_numで指定したクラスターのどこに割り振られたかの情報をテキストファイルに保存する際に、
クラスターでソートするやり方です。
このデータは、gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知なので、
「G1群で高発現DEGだとわかっているもの(gene_1〜gene_1800)がどのパターンに割り振られたか、gene_1801〜gene_2000がどのパターンに割り振られたか、
も表示させています。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge3.png"
out_f2 <- "hoge3.txt"
param_fig <- c(800, 500)
param_clust_num <- 4
param_G1 <- 3
param_G2 <- 3
library(MBCluster.Seq)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
table(cls$cluster)
table(cls$cluster[1:1800])
table(cls$cluster[1801:2000])
table(cls$cluster[2001:10000])
png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(data), normalized, cls$cluster)
tmp <- tmp[order(cls$cluster),]
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータ。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
入力ファイル以外は、3.と基本的に同じ。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge4.png"
out_f2 <- "hoge4.txt"
param_fig <- c(800, 500)
param_clust_num <- 5
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
library(MBCluster.Seq)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
table(cls$cluster)
png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(data), normalized, cls$cluster)
tmp <- tmp[order(cls$cluster),]
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
5. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
サンプルデータ15のシミュレーションデータ作成から行うやり方です。
混同行列(confusion matrix)情報のファイルも出力しています。
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5_confusion.txt"
param_clust_num <- 4
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
library(TCC)
library(MBCluster.Seq)
library(edgeR)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
hoge <- RNASeq.Data(data, Normalizer=log(size.factors),
Treatment=data.cl, GeneID=rownames(data))
head(hoge$Normalizer, n=1)
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
conversion <- 1:param_clust_num
names(conversion) <- 1:param_clust_num
pattern <- cls$cluster
hoge <- table(cls$cluster[nonDEG_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[1]
conversion[cls_num] <- param_narabi[1]
hoge <- table(cls$cluster[DEG_G1_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[2]
conversion[cls_num] <- param_narabi[2]
hoge <- table(cls$cluster[DEG_G2_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[3]
conversion[cls_num] <- param_narabi[3]
hoge <- table(cls$cluster[DEG_G3_posi])
cls_num <- as.integer(names(hoge)[hoge == max(hoge)])
pattern[pattern == cls_num] <- param_narabi[4]
conversion[cls_num] <- param_narabi[4]
confusion <- matrix(0, nrow=param_clust_num, ncol=param_clust_num)
for(i in 1:param_clust_num){
posi <- which(conversion == param_narabi[i])
confusion[i, 1] <- sum(cls$cluster[nonDEG_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 2] <- sum(cls$cluster[DEG_G1_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 3] <- sum(cls$cluster[DEG_G2_posi] == posi)
posi <- which(conversion == param_narabi[i])
confusion[i, 4] <- sum(cls$cluster[DEG_G3_posi] == posi)
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, cls$cluster, pattern)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
- MBCluster.Seq:Si et al., Bioinformatics, 2014
- k-means++:Arthur and Vassilvitskii, SODA '07 Proceedings of the eighteenth annual ACM-SIAM symposium on Discrete algorithms, 2007
- TCC:Sun et al., BMC Bioinformatics, 2013
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR中のGLM論文:McCarthy et al., Nucleic Acids Res., 2012
解析 | クラスタリング | scRNA-seq | について
single-cell RNA-seq (scRNA-seq)データ用です。scRNA-toolsはscRNA-seqデータ解析全般のツールのデータベースですが、
この中にクラスタリングプログラムも含まれています。(scRNA-seqとの対比として)昔ながらのbulk RNA-seqでは、どのサンプルがどの群に属するかが既知なので
群間で発現が異なる遺伝子(Differentially Expressed Genes; DEG)の検出がcommon taskでした。しかしscRNA-seqでは、通常どのサンプルがどの群に属するかが不明なので、
探索的な解析(Exploratory Analysis)が中心となります。つまりクラスタリングが重要だということです。そのため、多くのscRNA-seq用のクラスタリングプログラムは
どのサンプルがどの群に属するかを割り当てることにフォーカスしています(Zappia et al., Genome Biol., 2017)。
このやり方はサンプルが細胞の状態が変化しない成熟細胞(mature cells)の場合に有効です。しかし発生段階(developmental stages)では、幹細胞(stem cells)が成熟細胞へと分化します。
よって、特定の群への割り当てというのは適切ではなく、ある細胞(one cell type)が別の種類の細胞へと連続的に変化していく軌跡(continuous trajectory)を並べる(ordering)ようなプログラムを利用する必要があります。
bulk RNA-seqでも経時変化に特化したものと通常のクラスタリングに分けられるようなものだと思えば納得しやすいでしょう。
また、クラスタリングの際に重要なのは、効果的な次元削減(dimensionality reduction)です。例えば、主成分分析(PCA)だと数万遺伝子(つまり数万次元)のデータを2次元や3次元に削減した状態でデータを表示させています。
このPCA (Rのprcomp関数を実行することと同じ)もまた、次元削減法の1つと捉えることができます。scRNA-seqデータに特化させたのが、t-SNE, ZIFA, CIDRのような以下にリストアップされているものたちです。
UMAPやSPRINGは、いずれもForceAtlas2というforce-directed layout algorithmを採用しているようです(Luecken and Theis, Mol Syst Biol., 2019のpage 9の右下)。
クラスタリングのアプローチには、K-meansのような通常のクラスタリングアルゴリズムの他に、community detection methodsというものもあります。
これは、各細胞(each cell)を「K個の最も類似度の高い細胞(K most similar cells)」とのみ連結させてグラフを描くK-Nearest Neighbour approach (KNN graph)
であるため、他の全細胞との類似度を保持する通常の方法に比べて高速です(Luecken and Theis, Mol Syst Biol., 2019のpage 11の右下)。
PhenoGraphなどがこれに属します。
ScanpyやSeuratもLouvain algorithm on single-cell KNN graphsがデフォルトのクラスタリング法として実行されるようなので(Luecken and Theis, Mol Syst Biol., 2019のpage 12の左)、
これらもcommunity detection系ということでよいのだろうと思います。
「Louvainアルゴリズム」でググると、scRNA-seqそのものずばりではありませんが、一般的な説明があります。
感覚的には、偶然に近いものとして連結されるグループの期待値よりも多く連結されるグループを「コミュニティ(community)」として同定するものです。
KNNグラフの中から密に連結される部分グラフを探すアルゴリズム、と理解してもよいと思います。
このあたりではmodularity optimizationという用語が出てきますが、感覚的にはsignal-to-noize ratio (S/N比)が高いみたいなイメージで、
「グループ間の連結度合いは少なく、グループ内の連結度合いが高いものがよい」みたいな理解でよいと思います。
trajectory系については、全てのタイプのtrajectoriesに対しても有用な方法というのはないが(Luecken and Theis, Mol Syst Biol., 2019のpage 16の左上)、
分枝が多くないシンプルなモデルの場合にはslingshotがよく、
より複雑なモデルの場合はPAGAがよいという報告(Saelens et al., Nat Biotechnol., 2019)があります。
R用:
- Monocle(trajectory用):Trapnell et al., Nat Biotechnol., 2014
- Seurat(通常用;community detection系):Satija et al., Nat Biotechnol., 2015
- Sincell(trajectory用):Juliá et al., Bioinformatics, 2015
- MAST(通常用):Finak et al., Genome Biol., 2015
- TSCAN(trajectory用):Ji and Ji, Nucleic Acids Res., 2016
- Mpath(trajectory用):Chen et al., Nat Commun., 2016
- CellTree(trajectory用):duVerle et al., BMC Bioinformatics, 2016
- SC3(通常用):Kiselev et al., Nat Methods, 2017
- CIDR(通常用):Lin et al., Genome Biol., 2017
- Monocle 2(trajectory用):Qiu et al., Nat Methods, 2017
- SAIC(通常用):Yang et al., BMC Genomics, 2017
- DIMM-SC(通常用):Sun et al., Bioinformatics, 2018
- ZINB-WaVE(通常用):Risso et al., Nat Commun., 2018
- dropClust(通常用):Sinha et al., Nucleic Acids Res., 2018
- BEARscc(通常用; spike-insを利用):Severson et al., Nat Commun., 2018
- slingshot(trajectory用):Street et al., BMC Genomics, 2018
- umap:Becht et al., Nat Biotechnol., 2018
- SOUP(?用):Zhu et al., Proc Natl Acad Sci U S A., 2019
- BAMM-SC(通常用;multiple individuals対応):Sun et al., Nat Commun., 2019
- SSCC(通常用):Ren et al., Genomics Proteomics Bioinformatics, 2019
- Vision(通常用;MSigDBという文字あり。annotaionなので分類に近い?!詳細不明):DeTomaso et al., Nat Commun., 2019
- PHATE(通常用;R版はphateRという名前です):Moon et al., Nat Biotechnol., 2019
R以外:
- Wanderlust(trajectory用; Wishboneが後継?!):Bendall et al., Cell, 2014
- PhenoGraph(原著論文とリンク先違うががたぶんこれが正解;community detection系):Pierson and Yau, Genome Biol., 2015
- ZIFA(通常用):Pierson and Yau, Genome Biol., 2015
- Wishbonet(trajectory用):Setty et al., Nat Biotechnol., 2016
- Tybalt(通常用):Way and Greene, Pac Symp Biocomput., 2018
- SPRING(通常用):Weinreb et al., Bioinformatics, 2018
- SCANPY(trajectory用;community detection系):Wolf et al., Genome Biol., 2018
- scVDMC(通常用):Zhang et al., PLoS Comput Biol., 2018
- scvis(?用):Ding et al., Nat Commun., 2018
- GraphDDP(trajectory用):Costa et al., Nat Commun., 2018
- scVI(通常用):Lopez et al., Nat Methods, 2018
- UMAP(通常用):Becht et al., Nat Biotechnol., 2018
- VASC(?用):Wang and Gu, Genomics Proteomics Bioinformatics, 2018
- COAC(通常用):Peng et al., PLoS Comput Biol., 2019
- PAGA(両方):Wolf et al., Genome Biol., 2019
- STREAM(trajectory用):Chen et al., Nat Commun., 2019
- VPAC(?用):Chen et al., BMC Bioinformatics, 2019
- scVAE(?用):Grønbech et al., bioRxiv, 2018
- PHATE(通常用):Moon et al., bioRxiv, 2018
解析 | クラスタリング | scRNA-seq | サンプル間 | PHATE(Moon_2019)
PHATEのR版であるphateRパッケージを用いて
サンプル間クラスタリング(次元削減と可視化;dimensionality reduction and visualization)を行うやり方を示します。
手順は開発者らが提供しているマウス骨髄のscRNA-seqデータのチュートリアル
に従っています。このチュートリアルでは「10 cells以上で発現している遺伝子」や
「総UMIカウント数が1000 よりも大きい細胞」のフィルタリングを行っていますが、下記の例題で用いる細胞数に応じて適宜変更しています。
例題1-3の実行結果は毎回変わります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
ここでは6細胞(6 cells)のデータとみなして考えます。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
library(phateR)
library(ggplot2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- phate(data)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PHATE1, y=PHATE2), size=param_size)
dev.off()
例題1と基本的に同じですが、どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
library(phateR)
library(ggplot2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- phate(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PHATE1, y=PHATE2, color=as.factor(data.cl)), size=param_size)
dev.off()
例題2では既知のグループラベル情報を与えましたが、cluster_phate関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
ここでは、クラスター数k = 2としています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_kmeans <- 2
library(phateR)
library(ggplot2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- phate(data)
grouped <- cluster_phate(out, k=param_kmeans)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PHATE1, y=PHATE2, color=as.factor(grouped)), size=param_size)
dev.off()
4. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題2とは入力ファイルが異なる以外は基本的に同じです。どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "sample51.txt"
out_f <- "hoge4.png"
param_G1 <- 29
param_G2 <- 29
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
library(phateR)
library(ggplot2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data.cl <- data.cl[obj]
data <- library.size.normalize(data)
data <- sqrt(data)
out <- phate(data)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PHATE1, y=PHATE2, color=as.factor(data.cl)), size=param_size)
dev.off()
5. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題3とは入力ファイルが異なる以外は基本的に同じです。
例題4では既知のグループラベル情報を与えましたが、cluster_phate関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
ここでは、クラスター数k = 2としています。
in_f <- "sample51.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
param_kmeans <- 2
library(phateR)
library(ggplot2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- phate(data)
grouped <- cluster_phate(out, k=param_kmeans)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PHATE1, y=PHATE2, color=as.factor(grouped)), size=param_size)
dev.off()
解析 | クラスタリング | scRNA-seq | サンプル間 | umap(Becht_2018)
umapパッケージを用いて
サンプル間クラスタリング(次元削減と可視化;dimensionality reduction and visualization)を行うやり方を示します。
手順は開発者らが提供しているマウス骨髄のscRNA-seqデータのチュートリアル
に従っています。このチュートリアルでは「10 cells以上で発現している遺伝子」や
「総UMIカウント数が1000 よりも大きい細胞」のフィルタリングを行っていますが、下記の例題で用いる細胞数に応じて適宜変更しています。
現状ではサンプル数の少なさに起因する「エラー: umap: number of neighbors must be smaller than number of items」というエラーメッセージに遭遇します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
ここでは6細胞(6 cells)のデータとみなして考えます。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_knn <- 3
library(umap)
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
tmp <- umap.defaults
tmp$n_neighbors <- param_knn
out <- as.data.frame(umap(data, config=tmp)$layout)
colnames(out) <- c("UMAP1", "UMAP2")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=UMAP1, y=UMAP2), size=param_size)
dev.off()
例題1と基本的に同じですが、どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_knn <- 3
library(umap)
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
tmp <- umap.defaults
tmp$n_neighbors <- param_knn
out <- as.data.frame(umap(data, config=tmp)$layout)
colnames(out) <- c("UMAP1", "UMAP2")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=UMAP1, y=UMAP2), color=as.factor(data.cl), size=param_size)
dev.off()
例題2では既知のグループラベル情報を与えましたが、kmeans関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_kmeans <- 2
param_knn <- 3
library(umap)
library(cclust)
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
tmp <- umap.defaults
tmp$n_neighbors <- param_knn
out <- as.data.frame(umap(data, config=tmp)$layout)
colnames(out) <- c("UMAP1", "UMAP2")
#grouped <- cclust(x=data, centers=param_kmeans,
# iter.max=100, verbose=T, method="kmeans")
grouped <- kmeans(x=data, centers=param_kmeans, nstart=100)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=UMAP1, y=UMAP2), color=as.factor(grouped$cluster), size=param_size)
dev.off()
4. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題2とは入力ファイルが異なる以外は基本的に同じですが、サンプル数がそれなりにあるのでparam_knnをなくしてデフォルトにしています。どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "sample51.txt"
out_f <- "hoge4.png"
param_G1 <- 29
param_G2 <- 29
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
library(umap)
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data.cl <- data.cl[obj]
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(umap(data)$layout)
colnames(out) <- c("UMAP1", "UMAP2")
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=UMAP1, y=UMAP2), color=as.factor(data.cl), size=param_size)
dev.off()
5. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題3とは入力ファイルが異なる以外は基本的に同じですが、サンプル数がそれなりにあるのでparam_knnをなくしてデフォルトにしています。
kmeans関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
in_f <- "sample51.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
param_kmeans <- 2
library(umap)
library(cclust)
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(umap(data)$layout)
colnames(out) <- c("UMAP1", "UMAP2")
#grouped <- cclust(x=data, centers=param_kmeans,
# iter.max=100, verbose=T, method="kmeans")
grouped <- kmeans(x=data, centers=param_kmeans, nstart=100)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=UMAP1, y=UMAP2), color=as.factor(grouped$cluster), size=param_size)
dev.off()
解析 | クラスタリング | scRNA-seq | サンプル間 | t-SNE(van der Maaten_2008)
Rtsneパッケージを用いて
サンプル間クラスタリング(t分布型確率的近傍埋め込み法;t-Distributed Stochastic Neighbor Embedding; t-SNE;van der Maaten and Hintonm, 2008)を行うやり方を示します。
手順は開発者らが提供しているマウス骨髄のscRNA-seqデータのチュートリアル
に従っています。このチュートリアルでは「10 cells以上で発現している遺伝子」や
「総UMIカウント数が1000 よりも大きい細胞」のフィルタリングを行っていますが、下記の例題で用いる細胞数に応じて適宜変更しています。
param_perplexityのところでは、perplexityというパラメータ(デフォルトは30)を指定します。
「should not be bigger than 3 * perplexity < nrow(X)- 1」と書かれており、
例題1-3のような細胞数(=6)が非常に少ないデータの場合は3*perplexity < 6 - 1となるので、perplexity = 1とせねばなりません。
実行結果は毎回変わります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
ここでは6細胞(6 cells)のデータとみなして考えます。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_perplexity <- 1
library(ggplot2)
library(Rtsne)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(Rtsne(data, perplexity=param_perplexity)$Y)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=V1, y=V2), size=param_size)
dev.off()
例題1と基本的に同じですが、どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_perplexity <- 1
library(ggplot2)
library(Rtsne)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(Rtsne(data, perplexity=param_perplexity)$Y)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=V1, y=V2, color=as.factor(data.cl)), size=param_size)
dev.off()
例題2では既知のグループラベル情報を与えましたが、kmeans関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
ここでは、クラスター数k = 2としています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_kmeans <- 2
param_perplexity <- 1
library(cclust)
library(ggplot2)
library(Rtsne)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(Rtsne(data, perplexity=param_perplexity)$Y)
#grouped <- cclust(x=data, centers=param_kmeans,
# iter.max=100, verbose=T, method="kmeans")
grouped <- kmeans(x=data, centers=param_kmeans, nstart=100)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=V1, y=V2, color=as.factor(grouped$cluster)), size=param_size)
dev.off()
4. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題2とは入力ファイルが異なる以外は基本的に同じですが、サンプル数がそれなりにあるのでparam_perplexityを18にしています。
どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "sample51.txt"
out_f <- "hoge4.png"
param_G1 <- 29
param_G2 <- 29
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
param_perplexity <- 18
library(ggplot2)
library(Rtsne)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data.cl <- data.cl[obj]
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(Rtsne(data, perplexity=param_perplexity)$Y)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=V1, y=V2, color=as.factor(data.cl)), size=param_size)
dev.off()
5. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題3とは入力ファイルが異なる以外は基本的に同じですが、サンプル数がそれなりにあるのでparam_perplexityを18にしています。
kmeans関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
ここでは、クラスター数k = 2としています。
in_f <- "sample51.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
param_kmeans <- 2
param_perplexity <- 18
library(cclust)
library(ggplot2)
library(Rtsne)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(Rtsne(data, perplexity=param_perplexity)$Y)
#grouped <- cclust(x=data, centers=param_kmeans,
# iter.max=100, verbose=T, method="kmeans")
grouped <- kmeans(x=data, centers=param_kmeans, nstart=100)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=V1, y=V2, color=as.factor(grouped$cluster)), size=param_size)
dev.off()
解析 | クラスタリング | scRNA-seq | サンプル間 | 主成分分析(PCA)
statsパッケージのprcomp関数を用いて
サンプル間クラスタリング(主成分分析;Principal Component Analysis; PCA)を行うやり方を示します。
手順は開発者らが提供しているマウス骨髄のscRNA-seqデータのチュートリアル
に従っています。このチュートリアルでは「10 cells以上で発現している遺伝子」や
「総UMIカウント数が1000 よりも大きい細胞」のフィルタリングを行っていますが、下記の例題で用いる細胞数に応じて適宜変更しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
ここでは6細胞(6 cells)のデータとみなして考えます。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(prcomp(data)$x)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PC1, y=PC2), size=param_size)
dev.off()
例題1と基本的に同じですが、どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.png"
param_G1 <- 3
param_G2 <- 3
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(prcomp(data)$x)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PC1, y=PC2, color=as.factor(data.cl)), size=param_size)
dev.off()
例題2では既知のグループラベル情報を与えましたが、kmeans関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
ここでは、クラスター数k = 2としています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 3.5
param_kmeans <- 2
library(cclust)
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(prcomp(data)$x)
#grouped <- cclust(x=data, centers=param_kmeans,
# iter.max=100, verbose=T, method="kmeans")
grouped <- kmeans(x=data, centers=param_kmeans, nstart=100)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PC1, y=PC2, color=as.factor(grouped$cluster)), size=param_size)
dev.off()
4. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題2とは入力ファイルが異なる以外は基本的に同じです。どの細胞がどの群に属するかという既知のサンプルラベル情報を与えて色分けしています。
in_f <- "sample51.txt"
out_f <- "hoge4.png"
param_G1 <- 29
param_G2 <- 29
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data.cl <- data.cl[obj]
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(prcomp(data)$x)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PC1, y=PC2, color=as.factor(data.cl)), size=param_size)
dev.off()
5. サンプルデータ51の714 genes×58 samplesのカウントデータ(sample51.txt)の場合:
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
例題3とは入力ファイルが異なる以外は基本的に同じです。kmeans関数を用いたKmeans clusteringを独立して行うことで、実際にグループ化した結果で色分けするやり方です。
ここでは、クラスター数k = 2としています。
in_f <- "sample51.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_cell <- 3
param_count <- 1000
param_size <- 1.8
param_kmeans <- 2
library(cclust)
library(ggplot2)
library(phateR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
data <- t(data)
dim(data)
obj <- colSums(data > 0) > param_cell
data <- data[, obj]
dim(data)
obj <- rowSums(data) > param_count
data <- data[obj, ]
dim(data)
data <- library.size.normalize(data)
data <- sqrt(data)
out <- as.data.frame(prcomp(data)$x)
#grouped <- cclust(x=data, centers=param_kmeans,
# iter.max=100, verbose=T, method="kmeans")
grouped <- kmeans(x=data, centers=param_kmeans, nstart=100)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
ggplot(out) +
geom_point(aes(x=PC1, y=PC2, color=as.factor(grouped$cluster)), size=param_size)
dev.off()
解析 | クラスタリング | scRNA-seq | 参照情報あり | について
scRNA-seqは探索的な解析(Exploratory Analysis)が中心です。
「解析 | クラスタリング | scRNA-seq | について」では手元のデータのみを用いてクラスタリングを行うプログラムをリストアップしていますが、
情報の蓄積により、このサンプルの発現プロファイルは「既知のこのクラスター(or cell type or cell identify)に近い」といった外部の情報を使って評価することもできます。
この外部の参照(refer)できる情報を利用して、手元にあるデータのサンプルのアノテーション(既知クラスター情報の注釈付けを行うというふうに理解すればよい)を行う作業を
reference-based cluster annotationとかclustering based on reference dataとかreference-based clusteringなどと表現します
(Luecken and Theis, Mol Syst Biol., 2019のpage 14の右中央あたり)。
従来の手元のデータのみでクラスタリングを行う作業は、これとの対比でdata-driven approachと表現されます。
当然ながら、細胞のタイプ(cell types)や組成は実験条件によっても異なりますし、アノテーションされないサンプルも結構な割合であるとは思いますので、
ここでリストアップされているプログラムが従来のやり方にとって代わることは、(情報の蓄積によって役割は大きくなるとは思いますが...)ないと思います
(Luecken and Theis, Mol Syst Biol., 2019のpage 14の右中央あたりにも同様の記述あり)。
scmapの論文中では"projection"という表現が、
そしてGarnettの論文中では"classification"や"annotation"という表現が使われています。
ですので、ここでリストアップされているものたちは、"分類"を行ってくれるものという風に解釈してもよいと思います。
R以外:
- scmap(web version):Kiselev et al., Nat Methods, 2018
- CellAtlasSearch:Srivastava et al., Nucleic Acids Res., 2018
- CellFishing.jl:Sato et al., Genome Biol., 2019
- Garnett:Pliner et al., Nat Methods, 2019
- a Python framework(SAVER-Xと似た枠組みらしい):Mieth et al., Sci Rep., 2019
解析 | 外れサンプル検出 | について
外れサンプル検出を行うプログラムをリストアップします。
解析 | 発現変動 | について(2013年頃の記載事項で記念に残しているだけ)
ここでは、RNA-seqから得られるカウントデータの特徴や、実験デザイン、モデルなどについて基本的なところを述べています。
しかし、以下の記述内容は2012年頃までの知識に基づいているため結構古くなっています。参考程度にしてください。
2014年7月現在の印象としては、ReExpressのような新規転写物を同定しつつ、
既知転写物とそれらの新規転写物配列情報をアップデートしながら発現量推定を行い、
かつ発現変動解析まで行う、あるいはそれを繰り返すという方向性に移行しつつあるような感じです。
つまり、これまでの行数一定の"遺伝子"発現行列データを入力として解析するのではなく、
逐次転写物の発現量の数値や行数が変化しながら解析を行うようなイメージです。
カウントデータとモデル
私のマイクロアレイの論文(Kadota et al., 2009)では、「データ正規化法」と「発現変動遺伝子検出法」の組合せに相性がある、ということを述べています。NGSデータ解析時にも同様な注意が必要です。
ここでの入力データは「遺伝子発現行列」です。「raw countsのもの」か「正規化後」の遺伝子発現行列のどちらを入力データとして用いるのかは用いる方法によりますが、特に指定のない限り「raw countsのデータ」を入力として下さい。
NGSデータは測定値が何回sequenceされたかというcount情報に基づくもので、digital gene expression (DGE)データなどと呼ばれます。
そして、生のカウントデータは当然ながら、integer (整数;小数点以下の数値を含むはずがない)です。
ある遺伝子由来のsequenceされたリード数は二項分布(binomial distribution)に従うと様々な論文中で言及されています。つまり、
- 一般的な二項分布の説明:1(成功)か0(失敗)かのいずれかのn回の独立な試行を行ったときの1の数で表現される離散確率分布で、1が出る確率をpとしている。
- NGSに置き換えた説明:マップされたトータルのリード数がnで、あるリードに着目したときにある特定の遺伝子由来のものである確率がp、ということになります。
ポアソン分布(Poisson distribution)に従う、という記述もよくみかけますが本質的に同じ意味(ポアソン分布や正規分布は二項分布の近似)です。
つまり、nが大きいのがNGSの特徴であるので、pが小さくても、np(sequenceされたある特定の遺伝子由来のリード数の期待値、に相当; λ=np)がそこそこ大きな値になることが期待されるわけです。
というわけですので、このような二項分布を仮定したモデルに基づく「発現変動遺伝子検出法」を用いる際には、
へたに正規化を行ってそのモデルを仮定することができなくなるような墓穴を掘ることはないでしょう、ということです。
ちなみに「負の二項分布(negative bionomial distribution)」というキーワードを目にすることもあろうかと思いますが、これは二項分布の拡張版です。
いわゆるtechnical variation (技術的なばらつき)は「ポアソン分布」として考えることができるが、
biological variation (生物学的なばらつき;もう少し正確にはbiological replicatesのvariation)は一般にそれよりも大きいので(サンプル間の変動係数(coefficient of variation; CV)を)additional variationとして取り扱うべく「負の二項(Negative Binomial; NB)分布」として考える必要性があるのでしょうね。(Robinson and Oshlack, 2010)
technical replicatesとbiological replicatesの意味合い
technical replicatesとbiological replicatesの意味合いについて、もうすこしざっくりと説明します。
例えば「人間である私の体内の二つの組織間(例えば大脳(Brain; B)と皮膚(Skin; S))で発現変動している遺伝子を検出したい」という目的があったとします。
同じ組織でもばらつきを考慮する必要があるので大脳由来サンプルをいくつかに分割して(これがtechnical replicatesという意味)データを取得します。
皮膚由来サンプルについても同様です。これで得られたデータは(B1, B2, ...) vs. (S1, S2, ...)のようなreplicatesありのデータになります。
この私一個人のtechnical replicatesデータから発現変動遺伝子が得られたとします。
一般的に「私一個人内でのデータのばらつき(technical variation)」は「同じ人間だけど複数の別人の大脳(or 皮膚)由来サンプル間のばらつき(biological variation)」よりも小さいです。
ですので、technical replicates由来データで発現変動遺伝子(Differentially Expressed Genes; DEGs)を同定した結果は「そんなあほな!!」というくらいのDEGs数がレポートされます。
ここで注意しなければいけないのは、この結果は「私一個人の二つのサンプル間で発現変動している遺伝子がこれだけあった」ということを言っているだけで、
「(私が所属する)人間全般に言えることではない!」という点です。そしてもちろん一般には「私個人の事象についてではなく人間(ある特定の生物種)全般にいえる事象に興味がある」わけです。
それゆえ(biological replicatesのデータでないとユニバーサルな事象を理解するのが難しいために)ある程度理解が進んだ人はbiological replicatesが重要だと言っているわけです。
「モデルだ〜分布だ〜」ということと「ユーザーがプログラムを実行して得られる結果(発現変動遺伝子数や特徴)」との関係
「モデルだ〜分布だ〜」ということと「ユーザーがプログラムを実行して得られる結果(発現変動遺伝子数や特徴)」との関係についてですが、重要なのは「比較している二群間でどの程度差があれば発現変動していると判断するか?」です。
これをうまく表現するために、同一サンプルの(biological) replicatesデータから発現変動していないデータがどの程度ばらついているかをまず見積もります。
(このときにどういう数式でばらつきを見積もるのか?というところでいろんな選択肢が存在するがために沢山のRパッケージが存在しうるというわけですが、、、まあこれはおいといて)
負の二項分布モデル(negative binomial model; NB model)では分散VARをVAR = MEAN + φMEAN^2という数式(φ > 0)で表現するのが一般的です。
ちなみにポアソンモデルというのはNBモデルのφ = 0の場合(つまりVAR = MEAN; ある遺伝子の平均発現量がわかるとその遺伝子の分散が概ね平均と同じ、という意味)です。
計算手順としては、同一サンプルの(biological) replicatesデータからφがどの程度の値かを見積もります(推定します)。
ここまでで「ある発現変動していない遺伝子(non-DEGs)の平均発現量がこれくらいのときの分散はこれくらい」という情報が得られます。
これで発現変動していないものの分布(モデル)が決まります。その後、比較する二つのサンプルのカウント(発現量)データを一緒にして分散を計算し、
non-DEGsの分布のどの程度の位置にいるか(p値)を計算し、形式的には帰無仮説(Null Hypothesis)を棄却するかどうかという手順を経てDEG or non-DEGの判定をします。
用いる正規化法の違いが(non-DEGの)モデル構築にどう影響するか?どう関係するのか?
用いる正規化法の違いが(non-DEGの)モデル構築にどう影響するか?どう関係するのか?についてですが、これは極めてシンプルで、DEGかどうかの判定をする際に「比較する二つのサンプルのカウント(発現量)データを一緒にして分散を計算し...」を行う際に、サンプル間の正規化がうまくできていなければバラツキ(分散)を正しく計算できません。
ただそれだけですが、用いる正規化法次第で結果が大きく異なりうるのです(Robinson and Oshlack, 2010)。そして発現変動解析結果はその後の解析(GO解析など)にほとんどそのまま反映されますので私を含む多くのグループが正規化法の開発や性能評価といったあたりの研究を行っています。
正規化 | についてのところでも述べていますが、大抵の場合、
パッケージ独自の「正規化法」と「発現変動遺伝子検出法」が実装されており、「どの正規化法を使うか?」ということが「どの発現変動遺伝子検出法を使うか?」ということよりも結果に与える影響が大きい(Bullard et al., 2010; Kadota et al., 2012; Garmire and Subramanian, 2012)ことがわかりつつあります。
従来のサンプル間正規化法(RPMやTMM法)は比較する二群間(G1群 vs. G2群)でDEG数に大きな偏りがないことを前提にしています。
つまり、「(G1群 >> G2群)のDEG数と(G1群 << G2群)のDEG数が同程度」という前提条件がありますが、実際には大きく偏りがある場合も想定すべきで、偏りがあるなしにかかわらずロバストに正規化を行ってくれるのがTbT正規化法(Kadota et al., 2012)です。
偏りがあるなしの主な原因はDEGの分布にあるわけです。よって、TbT論文では、「正規化中にDEG検出を行い、DEGを除き、non-DEGのみで再度正規化係数を決める」というDEG elimination strategyに基づく正規化法を提案しています。
そして、TbT法を含むDEG elimination strategyに基づくいくつかの正規化法をTCCというRパッケージ中に実装しています(TCC論文中ではこの戦略をDEGES「でげす」と呼んでいます; Sun et al., 2013)。TCCパッケージ中に実装されている主な方法および入力データは以下の通りです:
- DEGES/TbT:オリジナルのTbT法そのもの。TMM-baySeq-TMMという3ステップで正規化を行う方法。step 2のDEG同定のところで計算時間のかかるbaySeqを採用しているため、他のパッケージでは数秒で終わる正規化ステップが数十分程度はかかるのがネック。また、biological replicatesがあることを前提としている。(A1, A2, ...) vs. (B1, B2, ...)のようなデータという意味。
- iDEGES/edgeR:DEGES/TbT法の超高速版(100倍程度高速)。TMM-(edgeR-TMM)nというステップで正規化を行う方法(デフォルトはn = 3)。step 2のDEG同定のところでedgeRパッケージ中のexact test (Robinson and Smyth, 2008)を採用しているためDEG同定の高速化が可能。
したがって、(step2 - step3)のサイクルをiterative (それゆえDEGESの頭にiterativeの意味を示すiがついたiDEGESとなっている)に行ってよりロバストな正規化係数を得ることが理論的に可能な方法です。
それゆえreplicatesのある二群間比較を行いたい場合にはDEGES/TbTよりもiDEGES/edgeRのほうがおすすめ。
- iDEGES/DESeq:一連のmulti-stepの正規化手順をすべてDESeqパッケージ中の関数のみで実装したパイプライン。複製実験のない二群間比較時(i.e., A1 vs. B1など)にはこちらを利用します。
TCCパッケージでは、内部的にedgeR, baySeq, DESeqを利用しています。
- an exact test:Robinson and Smyth, Biostatistics, 2008
- Kadota et al., Algorithms Mol., Biol., 2009
- Bullard et al., BMC Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- TbT正規化法:Kadota et al., Algorithms Mol., Biol., 2012
- Garmire and Subramanian, RNA, 2012
- TCC:Sun et al., BMC Bioinformatics, 2013
解析 | 発現変動 | RNA-seq | について
昔(2018年6月まで)は2群間用、3群間用などと別々にプログラムをリストアップしていましたが、もはやそんな時代ではありませんのでここに新規項目を作成しました。
scRNA-seq用については「解析 | 発現変動 | scRNA-seq | について」をご覧ください。
R用:
- DEGSeq:Wang et al., Bioinformatics, 2010
- edgeR:Robinson et al., Bioinformatics, 2010
- GPseq:Srivastava et al., Nucleic Acids Res., 2010
- baySeq:Hardcastle and Kelly, BMC Bioinformatics, 2010
- DESeq:Anders and Huber, Genome Biol., 2010
- ASC:Wu et al., BMC Bioinformatics, 2010
- NBPSeq:Di et al., SAGMB, 2011
- BBSeq:Zhou et al., Bioinformatics, 2011
- NOISeq:Tarazona et al., Genome Res., 2011
- PoissonSeq:Li et al., Biostatistics, 2012
- SAMseq (samr):Li and Tibshirani, Stat Methods Med Res., 2013
- BitSeq:Glaus et al., Bioinformatics, 2012
- easyRNASeq:Delhomme et al., Bioinformatics, 2012
- DSS:Wu et al., Biostatistics, 2012
- DSGseq:Wang et al., Gene, 2013
- EBSeq:Leng et al., Bioinformatics, 2013
- sSeq:Yu et al., Bioinformatics, 2013
- ALDEx:Fernandes et al., PLoS One, 2013
- TCC:Sun et al., BMC Bioinformatics, 2013
- tweeDEseq:Esnaola et al., BMC Bioinformatics, 2013
- NPEBseq:Bi et al., BMC Bioinformatics, 2013
- DER Finder:Frazee et al., Biostatistics, 2014
- voom (limma):Law et al., Genome Biol., 2014
- Characteristic Direction(CD):Clark et al., BMC Bioinformatics, 2014
- edgeR_robust:Zhou et al., Nucleic Acids Res., 2014
- ALDEx2:Fernandes et al., Microbiome, 2014
- ShrinkBayes:Van De Wiel et al., BMC Bioinformatics, 2014
- metaseqR:Moulos and Hatzis, Nucleic Acids Res., 2015
- DESeq2:Love et al., Genome Biol., 2014
- LFCseq:Lin et al., BMC Genomics, 2014
- rSeqNP:Shi et al., Bioinformatics, 2015
- edgeRun:Dimont et al., Bioinformatics, 2015
- deGPS:Chu et al., BMC Genomics, 2015
- contamDE:Shen et al., Bioinformatics, 2016
- derfinder:Collado-Torres et al., Nucleic Acids Res., 2016
- SeqMADE:Lei et al., Bioinformatics, 2017
- sleuth:Pimentel et al., Nat Methods, 2017
- CORNAS:Low et al., BMC Bioinformatics, 2017
- DREAMSeq:Gao et al., Front Genet., 2018
Review、ガイドライン、パイプライン系:
- プロトコル:Anders et al., Nat Protoc., 2013
- 手法比較(反復を増やすほうがdepthを増やすよりも効果的; Poisson-Seqとlimmaがよかった):Rapaport et al., Genome Biol., 2013
- 手法比較(DESeqが無難という結論):Seyednasrollah et al., Brief Bioinform., 2015
- 手法比較(edgeRがいいという結論):Guo et al., BMC Genomics, 2013
- compcodeR(benchmarking; 手法比較):Soneson C., Bioinformatics, 2014
- 手法比較(edgeRがいいという結論):Zhang et al., PLoS One, 2014
- 手法比較(DESeq2とedgeRがいいという結論):Ching et al., RNA, 2014
- 手法比較(edgeRがいいという結論; qPCR validation):Rajkumar et al., BMC Genomics, 2015
- 手法比較(Rapaport論文の結果はedgeRのnormalized data作成部分が間違っている。edgeRはいいという結論):Zhou and Robinson, Genome Biol., 2015
- 手法比較(qPCRとの相性の点ではGFOLDがよさそうという結論; triplicatesで十分だろう):Khang and Lau, PeerJ., 2015
- 手法比較(結論はよくわからない):Williams et al., BMC Bioinformatics, 2017
- 手法比較(様々な方法を組み合わせるとよい):Costa-Silva et al., PLoS One, 2017
- 手法比較(5反復など反復数が多い場合はALDEx2がよい):Quinn et al., BMC Bioinformatics, 2018
- 手法比較:Stupnikov et al., Comput Struct Biotechnol J., 2021
解析 | 発現変動 | 2群間 | 対応なし | について
実験デザインが以下のような場合にこのカテゴリーに属す方法を適用します:
Aさんの正常サンプル
Bさんの正常サンプル
Cさんの正常サンプル
Dさんの腫瘍サンプル
Eさんの腫瘍サンプル
Fさんの腫瘍サンプル
Gさんの腫瘍サンプル
2016年10月に調査した結果をリストアップします。PoissonSeqのリンク先がなかったので追加しました(2015/02/02)。
R用:
- DEGSeq:Wang et al., Bioinformatics, 2010
- edgeR:Robinson et al., Bioinformatics, 2010
- GPseq:Srivastava et al., Nucleic Acids Res., 2010
- baySeq:Hardcastle and Kelly, BMC Bioinformatics, 2010
- DESeq:Anders and Huber, Genome Biol., 2010
- ASC:Wu et al., BMC Bioinformatics, 2010
- NBPSeq:Di et al., SAGMB, 2011
- BBSeq:Zhou et al., Bioinformatics, 2011
- NOISeq:Tarazona et al., Genome Res., 2011
- PoissonSeq:Li et al., Biostatistics, 2012
- SAMseq (samr):Li and Tibshirani, Stat Methods Med Res., 2013
- BitSeq:Glaus et al., Bioinformatics, 2012
- easyRNASeq:Delhomme et al., Bioinformatics, 2012
- DSS:Wu et al., Biostatistics, 2012
- DSGseq:Wang et al., Gene, 2013
- EBSeq:Leng et al., Bioinformatics, 2013
- sSeq:Yu et al., Bioinformatics, 2013
- TCC:Sun et al., BMC Bioinformatics, 2013
- tweeDEseq:Esnaola et al., BMC Bioinformatics, 2013
- NPEBseq:Bi et al., BMC Bioinformatics, 2013
- DER Finder:Frazee et al., Biostatistics, 2014
- voom (limma):Law et al., Genome Biol., 2014
- Characteristic Direction(CD):Clark et al., BMC Bioinformatics, 2014
- edgeR_robust:Zhou et al., Nucleic Acids Res., 2014
- ShrinkBayes:Van De Wiel et al., BMC Bioinformatics, 2014
- metaseqR:Moulos and Hatzis, Nucleic Acids Res., 2015
- DESeq2:Love et al., Genome Biol., 2014
- LFCseq:Lin et al., BMC Genomics, 2014
- rSeqNP:Shi et al., Bioinformatics, 2015
- edgeRun:Dimont et al., Bioinformatics, 2015
- deGPS:Chu et al., BMC Genomics, 2015
- contamDE:Shen et al., Bioinformatics, 2016
- derfinder:Collado-Torres et al., Nucleic Acids Res., 2016
Review、ガイドライン、パイプライン系:
- プロトコル:Anders et al., Nat Protoc., 2013
- 手法比較(反復を増やすほうがdepthを増やすよりも効果的; Poisson-Seqとlimmaがよかった):Rapaport et al., Genome Biol., 2013
- 手法比較(DESeqが無難という結論):Seyednasrollah et al., Brief Bioinform., 2015
- 手法比較(edgeRがいいという結論):Guo et al., BMC Genomics, 2013
- compcodeR(benchmarking; 手法比較):Soneson C., Bioinformatics, 2014
- 手法比較(edgeRがいいという結論):Zhang et al., PLoS One, 2014
- 手法比較(DESeq2とedgeRがいいという結論):Ching et al., RNA, 2014
- 手法比較(edgeRがいいという結論; qPCR validation):Rajkumar et al., BMC Genomics, 2015
- 手法比較(Rapaport論文の結果はedgeRのnormalized data作成部分が間違っている。edgeRはいいという結論):Zhou and Robinson, Genome Biol., 2015
- 手法比較(qPCRとの相性の点ではGFOLDがよさそうという結論; triplicatesで十分だろう):Khang and Lau, PeerJ. , 2015
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | DESeq2(Love_2014)
DESeq2パッケージ (Love et al., Genome Biol., 2014)を用いるやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge1.txt"
out_f2 <- "hoge1.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(400, 380)
library(DESeq2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
#d <- estimateSizeFactors(d)
#d <- estimateDispersions(d)
#d <- nbinomLRT(d, full= ~condition, reduced= ~1)
d <- DESeq(d)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
sum(q.value < param_FDR)
sum(p.adjust(p.value, method="BH") < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotMA(d)
dev.off()
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | TCC(Sun_2013)
TCCを用いたやり方を示します。
内部的にiDEGES/edgeR(Sun_2013)正規化を実行したのち、
edgeRパッケージ中のexact testで発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行っています。TCC原著論文中のiDEGES/edgeR-edgeRという解析パイプラインに相当します。
全てTCCパッケージ(Sun et al., BMC Bioinformatics, 2013)内で完結します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge1.txt"
out_f2 <- "hoge1.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-result$rank))
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
正規化後のテキストファイルデータを出力し、平均-分散プロットのpngファイルを出力しています。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge2.txt"
out_f2 <- "hoge2.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(380, 420)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
hoge <- normalized
MEAN <- apply(hoge, 1, mean)
VARIANCE <- apply(hoge, 1, var)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(MEAN, VARIANCE, log="xy", pch=20, cex=.1,
xlim=c(1e-02, 1e+08), ylim=c(1e-02, 1e+08), col="black")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
obj <- (result$q.value < param_FDR)
points(MEAN[obj], VARIANCE[obj], col="magenta", cex=0.1, pch=20)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
DEG検出後に任意のFDR閾値を満たす遺伝子数を調べたり、色分けしたりx, y軸の範囲を限定するなどする様々なテクニックを示しています。
in_f <- "data_hypodata_3vs3.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.1
param_nonDEG <- 2001:10000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
sum(tcc$estimatedDEG)
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
sum(tcc$DEGES$potentialDEG)
tcc$norm.factors
out_f1 <- "hoge3_all_010.png"
param_fig <- c(400, 380)
M <- getResult(tcc)$m.value
A <- getResult(tcc)$a.value
png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)",
xlim=c(-2, 16), ylim=c(-8, 6), pch=20, cex=.1)
grid(col="gray", lty="dotted")
obj <- tcc$stat$q.value < param_FDR
points(A[obj], M[obj], col="magenta", cex=0.1, pch=20)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(M[obj] < 0)
sum(M[obj] > 0)
tcc <- tcc[param_nonDEG]
out_f2 <- "hoge3_nonDEG_010.png"
M <- getResult(tcc)$m.value
A <- getResult(tcc)$a.value
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)",
xlim=c(-2, 16), ylim=c(-8, 6), pch=20, cex=.1)
grid(col="gray", lty="dotted")
obj <- tcc$stat$q.value < param_FDR
points(A[obj], M[obj], col="magenta", cex=0.1, pch=20)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(M[obj] < 0)
sum(M[obj] > 0)
param_FDR <- 0.2
out_f3 <- "hoge3_nonDEG_020.png"
M <- getResult(tcc)$m.value
A <- getResult(tcc)$a.value
png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)",
xlim=c(-2, 16), ylim=c(-8, 6), pch=20, cex=.1)
grid(col="gray", lty="dotted")
obj <- tcc$stat$q.value < param_FDR
points(A[obj], M[obj], col="magenta", cex=0.1, pch=20)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(M[obj] < 0)
sum(M[obj] > 0)
param_FDR <- 0.4
out_f4 <- "hoge3_nonDEG_040.png"
M <- getResult(tcc)$m.value
A <- getResult(tcc)$a.value
png(out_f4, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)",
xlim=c(-2, 16), ylim=c(-8, 6), pch=20, cex=.1)
grid(col="gray", lty="dotted")
obj <- tcc$stat$q.value < param_FDR
points(A[obj], M[obj], col="magenta", cex=0.1, pch=20)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(M[obj] < 0)
sum(M[obj] > 0)
param_FDR <- 0.6
out_f5 <- "hoge3_nonDEG_060.png"
M <- getResult(tcc)$m.value
A <- getResult(tcc)$a.value
png(out_f5, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(A, M, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)",
xlim=c(-2, 16), ylim=c(-8, 6), pch=20, cex=.1)
grid(col="gray", lty="dotted")
obj <- tcc$stat$q.value < param_FDR
points(A[obj], M[obj], col="magenta", cex=0.1, pch=20)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(M[obj] < 0)
sum(M[obj] > 0)
4. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の場合:
「FDR閾値を満たすもの」と「fold-change閾値を満たすもの」それぞれのM-A plotを作成しています。
in_f <- "data_marioni.txt"
out_f1 <- "hoge4.txt"
out_f2 <- "hoge4_FDR.png"
out_f3 <- "hoge4_FC.png"
param_G1 <- 5
param_G2 <- 5
param_FDR <- 0.05
param_FC <- 2
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
M <- getResult(tcc)$m.value
hoge <- rep(1, length(M))
hoge[abs(M) > log2(param_FC)] <- 2
cols <- c("black", "magenta")
png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, col=cols, col.tag=hoge, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(", param_FC, "-fold)", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(abs(M) > log2(16))
sum(abs(M) > log2(8))
sum(abs(M) > log2(4))
sum(abs(M) > log2(2))
5. サンプルデータ4の18,110 genes×10 samplesのリアルデータ(data_marioni.txt; kidney 5サンプル vs. liver 5サンプル)の最初の4サンプルを比較する場合:
「FDR閾値を満たすもの」と「fold-change閾値を満たすもの」それぞれのM-A plotを作成しています。
in_f <- "data_marioni.txt"
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5_FDR.png"
out_f3 <- "hoge5_FC.png"
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_FC <- 2
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,1:(param_G1+param_G2)]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
M <- getResult(tcc)$m.value
hoge <- rep(1, length(M))
hoge[abs(M) > log2(param_FC)] <- 2
cols <- c("black", "magenta")
png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, col=cols, col.tag=hoge, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(", param_FC, "-fold)", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(abs(M) > log2(16))
sum(abs(M) > log2(8))
sum(abs(M) > log2(4))
sum(abs(M) > log2(2))
6. サンプルデータ8の26,221 genes×6 samplesのリアルデータ(data_arab.txt; mock 3サンプル vs. hrcc 3サンプル)の場合:
「FDR閾値を満たすもの」と「fold-change閾値を満たすもの」それぞれのM-A plotを作成しています。
in_f <- "data_arab.txt"
out_f1 <- "hoge6.txt"
out_f2 <- "hoge6_FDR.png"
out_f3 <- "hoge6_FC.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_FC <- 2
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,1:(param_G1+param_G2)]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
M <- getResult(tcc)$m.value
hoge <- rep(1, length(M))
hoge[abs(M) > log2(param_FC)] <- 2
cols <- c("black", "magenta")
png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, col=cols, col.tag=hoge, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(", param_FC, "-fold)", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(abs(M) > log2(16))
sum(abs(M) > log2(8))
sum(abs(M) > log2(4))
sum(abs(M) > log2(2))
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
1.と基本的に同じで、出力のテキストファイルが正規化前のデータではなく正規化後のデータになっていて、発現変動順にソートしたものになっています。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
Neyret-Kahn et al., Genome Res., 2013のgene-levelの2群間比較用(3 proliferative samples vs. 3 Ras samples)ヒトRNA-seqカウントデータです。
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)から得られます。
in_f <- "hoge9_count_gene.txt"
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(430, 390)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
8.の入力ファイル(hoge9_count_gene.txt)と本質的に同じもの(アノテーション情報が2014年3月ごろと若干古いだけ)です。
パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)から得られます。
in_f <- "srp017142_count_bowtie.txt"
out_f1 <- "hoge9.txt"
out_f2 <- "hoge9.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(430, 390)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR, xlim=c(-3, 17), ylim=c(-8, 15))
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
- TCC:Sun et al., BMC Bioinformatics, 2013
- TbT正規化法(TCCに実装されたDEGESアルゴリズム提唱論文):Kadota et al., Algorithms Mol. Biol., 2012
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- an exact test for negative binomial distribution:Robinson and Smyth, Biostatistics, 2008
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | Blekhmanデータ | TCC(Sun_2013)
Blekhman et al., Genome Res., 2010の公共カウントデータ解析に特化させて、
TCCを用いた様々な例題を示します。
入力は全てサンプルデータ42の20,689 genes×18 samplesのリアルカウントデータ
(sample_blekhman_18.txt)です。
ヒトHomo sapiens; HS)のメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3),
チンパンジー(Pan troglodytes; PT)のメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザル(Rhesus macaque; RM)のメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
つまり、以下のような感じです。FはFemale(メス)、MはMale(オス)を表します。
ヒト(1-6列目): HSF1, HSF2, HSF3, HSM1, HSM2, and HSM3
チンパンジー(7-12列目): PTF1, PTF2, PTF3, PTM1, PTM2, and PTM3
アカゲザル(13-18列目): RMF1, RMF2, RMF3, RMM1, RMM2, and RMM3
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
1, 4, 13, 16
列目のデータのみ抽出しています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge1.txt"
out_f2 <- "hoge1.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10, 10),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
2. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
1.と基本的に同じで、ランキング1位の"ENSG00000208570"をハイライトさせるテクニックです。
1位のy軸の値(m.value)が11.29なので、y軸の範囲を[-10.0, 11.5]の範囲に変更する操作も行っています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge2.txt"
out_f2 <- "hoge2.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(400, 310)
param_mar <- c(4, 4, 0, 0)
param_geneid <- "ENSG00000208570"
param_col <- "magenta"
param_cex <- 2
param_pch <- 20
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
obj <- is.element(tcc$gene_id, param_geneid)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
3. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
2.と基本的に同じです。
ランキング2位の"ENSG00000220191"をハイライトさせるテクニックです。
param_cexやparam_pchを変更しています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge3.txt"
out_f2 <- "hoge3.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(400, 310)
param_mar <- c(4, 4, 0, 0)
param_geneid <- "ENSG00000220191"
param_col <- "magenta"
param_cex <- 2.3
param_pch <- 15
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
obj <- is.element(tcc$gene_id, param_geneid)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
4. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
3.と基本的に同じです。
ランキング3位の"ENSG00000106366"をハイライトさせるテクニックです。
param_cexやparam_pchを変更しています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge4.txt"
out_f2 <- "hoge4.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(400, 310)
param_mar <- c(4, 4, 0, 0)
param_geneid <- "ENSG00000106366"
param_col <- "magenta"
param_cex <- 3.1
param_pch <- 18
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
obj <- is.element(tcc$gene_id, param_geneid)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
5. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
4.と基本的に同じです。
param_FDRで指定した閾値をギリギリ満たすランキング2,488位の"ENSG00000139445"をハイライトさせるテクニックです。
param_col, param_cex, param_pchを変更しています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(400, 310)
param_mar <- c(4, 4, 0, 0)
param_geneid <- "ENSG00000139445"
param_col <- "skyblue"
param_cex <- 2.5
param_pch <- 17
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
obj <- is.element(tcc$gene_id, param_geneid)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
6. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
3種類のFDR閾値(0.0001, 0.05, 0.4)でのM-A plotを出力させています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge6_1.png"
out_f2 <- "hoge6_2.png"
out_f3 <- "hoge6_3.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- c(0.0001, 0.05, 0.4)
param_fig <- c(375, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger")
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR[1])
sum(tcc$stat$q.value < param_FDR[2])
sum(tcc$stat$q.value < param_FDR[3])
png(out_f1, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR[1], xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR[1], ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR[2], xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR[2], ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR[3], xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR[3], ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
ここでは、1, 4, 7, 10
列目のデータのみ抽出して、ヒト2サンプル(G1群:HSF1とHSM1) vs. チンパンジー2サンプル(G2群:PTF1とPTM1)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7.png"
param_subset <- c(1, 4, 7, 10)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
colnames(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
ここでは、1, 4, 2, 5
列目のデータのみ抽出して、ヒト2サンプル(G1群:HSF1とHSM1) vs. ヒト2サンプル(G2群:HSF2とHSM2)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8.png"
param_subset <- c(1, 4, 2, 5)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
colnames(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
obj <- as.logical(tcc$stat$q.value < param_FDR)
points(result$a.value[obj], result$m.value[obj],
pch=20, cex=1.5, col="magenta")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
ここでは、7, 10, 8, 11列目のデータのみ抽出して、
チンパンジー2サンプル(G1群:PTF1とPTM1) vs. チンパンジー2サンプル(G2群:PTF2とPTM2)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge9.txt"
out_f2 <- "hoge9.png"
param_subset <- c(7, 10, 8, 11)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
colnames(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
obj <- as.logical(tcc$stat$q.value < param_FDR)
points(result$a.value[obj], result$m.value[obj],
pch=20, cex=1.5, col="magenta")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
ここでは、13, 16, 14, 17列目のデータのみ抽出して、
アカゲザル2サンプル(G1群:RMF1とRMM1) vs. アカゲザル2サンプル(G2群:RMF2とRMM2)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge10.txt"
out_f2 <- "hoge10.png"
param_subset <- c(13, 16, 14, 17)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
colnames(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
obj <- as.logical(tcc$stat$q.value < param_FDR)
points(result$a.value[obj], result$m.value[obj],
pch=20, cex=1.5, col="magenta")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
ここでは、13, 16, 15, 18列目のデータのみ抽出して、
アカゲザル2サンプル(G1群:RMF1とRMM1) vs. アカゲザル2サンプル(G2群:RMF3とRMM3)の2群間比較を行います。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge11.txt"
out_f2 <- "hoge11.png"
param_subset <- c(13, 16, 15, 18)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
colnames(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
obj <- as.logical(tcc$stat$q.value < param_FDR)
points(result$a.value[obj], result$m.value[obj],
pch=20, cex=1.5, col="magenta")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
12. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
2.と基本的に同じで、q.value = 1の"ENSG00000125844"と
"ENSG00000115325"をハイライトさせるテクニックです。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge12.txt"
out_f2 <- "hoge12.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(400, 310)
param_mar <- c(4, 4, 0, 0)
param_geneid <- c("ENSG00000125844", "ENSG00000115325")
param_col <- "skyblue"
param_cex <- 2
param_pch <- 20
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
obj <- is.element(tcc$gene_id, param_geneid)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
13. ヒト2サンプル(G1群:HSF1とHSM1) vs. アカゲザル2サンプル(G2群:RMF1とRMM1)の場合:
1.と基本的に同じで、「FDR閾値を満たすもの」と「fold-change閾値を満たすもの」それぞれのM-A plotを作成しています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge13.txt"
out_f2 <- "hoge13_FDR.png"
out_f3 <- "hoge13_FC.png"
param_subset <- c(1, 4, 13, 16)
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_FC <- 2
param_fig <- c(400, 310)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
M <- getResult(tcc)$m.value
hoge <- rep(1, length(M))
hoge[abs(M) > log2(param_FC)] <- 2
cols <- c("black", "magenta")
png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, col=cols, col.tag=hoge, xlim=c(-2, 17), ylim=c(-10.0, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(", param_FC, "-fold)", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(abs(M) > log2(16))
sum(abs(M) > log2(8))
sum(abs(M) > log2(4))
sum(abs(M) > log2(2))
- Blekhman et al., Genome Res., 2010
- TCC:Sun et al., BMC Bioinformatics, 2013
- TbT正規化法(TCCに実装されたDEGESアルゴリズム提唱論文):Kadota et al., Algorithms Mol. Biol., 2012
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- an exact test for negative binomial distribution:Robinson and Smyth, Biostatistics, 2008
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | SAMseq(Li_2013)
samrパッケージ中のSAMseq (Li and Tibshirani, 2013)を用いて
発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
library(samr)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
out <- SAMseq(data, data.cl, resp.type="Two class unpaired")
p.value <- samr.pvalues.from.perms(out$samr.obj$tt, out$samr.obj$ttstar)
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < 0.10)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
1.と基本的に同じで、DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_DEG <- 1:2000
library(samr)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
out <- SAMseq(data, data.cl, resp.type="Two class unpaired")
p.value <- samr.pvalues.from.perms(out$samr.obj$tt, out$samr.obj$ttstar)
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < 0.10)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
3. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータの作成から行う場合:
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3」の計6サンプル分からなります。
全10,000遺伝子中の最初の2,000個(gene_1〜gene_2000まで)が発現変動遺伝子(DEG)です。
全2,000 DEGsの内訳:最初の90%分(gene_1〜gene_1800)がG1群で4倍高発現、残りの10%分(gene_1801〜gene_2000)がG2群で4倍高発現
このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
out_f <- "hoge3.txt"
library(TCC)
library(samr)
library(ROC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=10000, PDEG=0.2,
DEG.assign=c(0.9, 0.1),
DEG.foldchange=c(4, 4),
replicates=c(3, 3))
data <- tcc$count
data.cl <- tcc$group$group
out <- SAMseq(data, data.cl, resp.type="Two class unpaired")
p.value <- samr.pvalues.from.perms(out$samr.obj$tt, out$samr.obj$ttstar)
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < 0.10)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
4. サンプルデータ13と同じ10,000 genes×6 samplesのシミュレーションデータの作成から行う場合:
「G1_rep1, G1_rep2, G1_rep3, G2_rep1, G2_rep2, G2_rep3」の計6サンプル分からなります。
全10,000遺伝子中の最初の2,000個(gene_1〜gene_2000まで)が発現変動遺伝子(DEG)です。
全2,000 DEGsの内訳:最初の90%分(gene_1〜gene_1800)がG1群で4倍高発現、残りの10%分(gene_1801〜gene_2000)がG2群で4倍高発現
このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
samrパッケージではなくTCCパッケージを用いています。
out_f <- "hoge4.txt"
library(TCC)
library(samr)
library(ROC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=10000, PDEG=0.2,
DEG.assign=c(0.9, 0.1),
DEG.foldchange=c(4, 4),
replicates=c(3, 3))
tcc <- estimateDE(tcc, test.method="samseq")
result <- getResult(tcc, sort=FALSE)
sum(q.value < 0.10)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | edgeR(Robinson_2010)
edgeRパッケージを用いて発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描くやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plotSmear(d)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描き、FDR < 0.05を満たすものを赤色で示すやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
hoge <- rownames(data)[q.value < param_FDR]
plotSmear(d, de.tags=hoge)
length(hoge)
length(hoge)/nrow(data)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描き、FDR値で発現変動順に並べた上位300個を赤色で示すやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge4.txt"
param_G1 <- 3
param_G2 <- 3
param3 <- 300
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
hoge <- rownames(data)[ranking <= param3]
plotSmear(d, de.tags=hoge)
length(hoge)
length(hoge)/nrow(data)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描き、2倍以上発現変化しているものを赤色で示すやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param3 <- 2
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
hoge <- rownames(data)[abs(out$table$logFC) >= log2(param3)]
plotSmear(d, de.tags=hoge)
length(hoge)
length(hoge)/nrow(data)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描き、MA-plotで大きさを指定してpng形式ファイルに保存するやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge6.txt"
out_f2 <- "hoge6.png"
param_G1 <- 3
param_G2 <- 3
param3 <- 2
param_fig <- c(600, 400)
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
hoge <- rownames(data)[abs(out$table$logFC) >= log2(param3)]
plotSmear(d, de.tags=hoge)
dev.off()
length(hoge)
length(hoge)/nrow(data)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描き、FDR < 0.01を満たすものを赤色で示したMA-plotをファイルに保存するやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.01
param_fig <- c(600, 400)
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
#write.table(tmp[order(ranking),], out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
hoge <- rownames(data)[q.value < param_FDR]
plotSmear(d, de.tags=hoge)
dev.off()
length(hoge)
length(hoge)/nrow(data)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描き、FDR < 0.01を満たすものを赤色で示したMA-plotをファイルに保存するやり方です。
基本は7と同じで、MA-plotの描画をplotSmear関数を用いないで行うやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.01
param_fig <- c(600, 400)
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
norm_f_RPM <- 1000000/colSums(data)
RPM <- sweep(data, 2, norm_f_RPM, "*")
data <- RPM
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
DEG_posi <- (q.value < param_FDR)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A=(log2(G2)+log2(G1))/2", ylab="M=log2(G2)-log2(G1)", pch=20, cex=.1)
grid(col="gray", lty="dotted")
points(x_axis[DEG_posi], y_axis[DEG_posi], col="red", pch=20, cex=0.1)
dev.off()
sum(DEG_posi)
sum(DEG_posi)/nrow(data)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
MA-plotも描き、FDR < 0.01を満たすものを赤色で示したMA-plotをファイルに保存するやり方です。
基本は8と同じで、MA-plotの描画をRPMではなくTMM正規化後のデータで行うやり方です。
in_f <- "data_hypodata_3vs3.txt"
out_f1 <- "hoge9.txt"
out_f2 <- "hoge9.png"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.01
param_fig <- c(600, 400)
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
group <- factor(data.cl)
d <- DGEList(counts=data,group=group)
keep <- filterByExpr(d)
#d <- d[keep,,keep.lib.sizes=FALSE]
d <- calcNormFactors(d)
design <- model.matrix(~group)
d <- estimateDisp(d, design)
fit <- glmQLFit(d, design)
out <- glmQLFTest(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
d <- DGEList(counts=data,group=data.cl)
d <- calcNormFactors(d)
norm_f_TMM <- d$samples$norm.factors
names(norm_f_TMM) <- colnames(data)
effective_libsizes <- colSums(data) * norm_f_TMM
RPM_TMM <- sweep(data, 2, 1000000/effective_libsizes, "*")
data <- RPM_TMM
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
DEG_posi <- (stat_edgeR < param_FDR)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A=(log2(G2)+log2(G1))/2", ylab="M=log2(G2)-log2(G1)", pch=20, cex=.1)
grid(col="gray", lty="dotted")
points(x_axis[DEG_posi], y_axis[DEG_posi], col="red", pch=20, cex=0.1)
dev.off()
sum(DEG_posi)
sum(DEG_posi)/nrow(data)
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- an exact test for negative binomial distribution:Robinson and Smyth, Biostatistics, 2008
- Robinson and Smyth, Bioinformatics, 2007
- McCarthy et al., Nucleic Acids Res., 2012
- edgeRをstem cell biologyに適用したという論文(QuasRを引用している):Nikolayeva and Robinson, Methods Mol Biol., 2014
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | WAD(Kadota_2008)
TCCパッケージを用いてiDEGES/edgeR正規化を行ったデータを入力として、
WAD法(Kadota et al., 2008)を適用して発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
WAD法は統計的手法ではない(ヒューリスティックな方法)ので、出力ファイル中にp-valueやq-valueは存在しません。
それゆえ、FDR閾値を満たす遺伝子数、という概念もありません。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="wad")
statistic <- tcc$stat$testStat
ranking <- tcc$stat$rank
tmp <- cbind(rownames(tcc$count), tcc$count, statistic, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
1.と基本的に同じで、DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_DEG <- 1:2000
library(TCC)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="wad")
statistic <- tcc$stat$testStat
ranking <- tcc$stat$rank
tmp <- cbind(rownames(tcc$count), tcc$count, statistic, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
2.と基本的に同じで、iDEGES/edgeR正規化後のデータを明示的に取得して、
estimateDE関数ではなくTCCパッケージ中のWAD関数を用いてWAD法を実行するやり方です。
WAD法はlog2変換後のデータを入力とすることを前提としており、発現レベルに相当する数値が1未満のものを1に変換してからlogをとっています。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 3
param_G2 <- 3
param_DEG <- 1:2000
library(TCC)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
head(normalized)
out <- WAD(normalized, data.cl, logged=F, floor=1)
ranking <- out$rank
tmp <- cbind(rownames(tcc$count), tcc$count, out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | varSelRF(Diaz-Uriarte_2007)
varSelRFパッケージを用いて2群の分類。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し、以下をコピペ
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3.txt"
param_G1 <- 3
param_G2 <- 3
library(varSelRF)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
out <- varSelRF(t(data), factor(data.cl))
out$selected.vars
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | について
以下でも記載していますが、
TCCは複製なしデータの場合には内部的にDESeq2をこれまで利用してきました。
しかしDESeq2が2018年10月頃リリース版で非対応となるらしいので、TCCもDESeqに切り替える予定です。
R用:
- edgeR:Robinson et al., Bioinformatics, 2010
- DESeq:Anders and Huber, Genome Biol., 2010
- ASC:Wu et al., BMC Bioinformatics, 2010
- NOISeq:Tarazona et al., Genome Res., 2011
- TCC:Sun et al., BMC Bioinformatics, 2013
- DESeq2(2018年10月頃リリース版で非対応となるらしい):Love et al., Genome Biol., 2014
- LPEseq:Gim et al., PLoS One, 2016
- CORNAS:Low et al., BMC Bioinformatics, 2017
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | CORNAS(Low_2017)
CORNASを用いるやり方を示します。
結構設定が面倒ですので、とりあえずCORNASの例題を示します。
githubは応答がないなどとよく言われて取り扱いづらいので、一旦全部ダウンロードしたものを改めてこのサイト上で提供しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Marioni et al., Genome Res., 2008のkidney vs. liverのカウントデータです。
configファイル(cornas.config.test4)も入力として利用します。
2018年7月19日現在、outオブジェクトにcornas関数実行結果がうまく格納されていません...。
in_f1 <- "test4_kidneyliver_example.tab"
in_f2 <- "cornas.config.test4"
out_f <- "hoge1.txt"
source("http://www.iu.a.u-tokyo.ac.jp/~kadota/R_seq/CORNAS-master/CORNAS.R")
out <- cornas(in_f2, in_f1)
write.table(out, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | LPEseq(Gim_2016)
LPEseqを用いるやり方を示します。
It is freely downloadable from our website (...) or Bioconductor.と7ページ目に書かれていますが、2018年7月現在Bioconductorでは提供されていません。
パッケージのインストールがまだの場合は、最初に例題0のやり方に従ってインストールをしておく必要があります。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
0. パッケージのインストール:
最新版のファイル(2018年12月現在はLPEseq_v0.99.1.tar.gz)を
作業ディレクトリ上に予めダウンロードしておいてから、下記をコピペしてください。
source("http://statgen.snu.ac.kr/software/LPEseq/LPEseq.R")
install.packages("LPEseq_v0.99.1.tar.gz", repos=NULL, type="source")
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
5% FDR閾値を満たす遺伝子数は2,317個、AUCは0.6397909であることがわかります。
in_f <- "data_hypodata_1vs1.txt"
out_f1 <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(LPEseq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
normalized <- LPEseq.normalise(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
G1 <- normalized[, data.cl==1]
G2 <- normalized[, data.cl==2]
out <- LPEseq.test(G1, G2, d=1)
p.value <- out$p.value
p.value[is.na(p.value)] <- 1
q.value <- out$q.value
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | NOISeq(Tarazona_2015)
NOISeqを用いるやり方を示します。
Tarazona et al., Genome Res., 2011が原著論文ですが、2015年の論文もあったのでそちらにしています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
param_thresは(1 - p-value)ではないとマニュアル中で強調されています。
この閾値を満たす遺伝子数は1,834個、AUC = 0.7811677でした。
in_f <- "data_hypodata_1vs1.txt"
out_f1 <- "hoge1.txt"
out_f2 <- "hoge1.png"
param_G1 <- 1
param_G2 <- 1
param_thres <- 0.9
param_fig <- c(400, 380)
library(NOISeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
myfactors <- data.frame(comp=data.cl)
eset <- readData(data=data, factors=myfactors)
out <- noiseq(eset, factor="comp",
k=NULL, norm="tmm", pnr=0.2, nss=5,
v=0.02, lc=1, replicates="no")
prob <- out@results[[1]]$prob
prob[is.na(prob)] <- 0
ranking <- rank(-prob)
sum(prob > param_thres)
hoge <- degenes(out, q=param_thres, M=NULL)
tmp <- cbind(rownames(data), data, prob, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | DESeq2(Love_2014)
DESeq2パッケージ
(Love et al., Genome Biol., 2014)を用いるやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
DESeq2パッケージ中の記述方式です
plotMAのところでエラーが出てM-A plotは描画できません(DESeq2 ver. 1.12.0で2016年5月22日に確認)。
in_f <- "data_hypodata_1vs1.txt"
out_f1 <- "hoge1.txt"
out_f2 <- "hoge1.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(DESeq2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
d <- DESeq(d)
#d <- estimateSizeFactors(d)
##sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))
#d <- estimateDispersions(d)
#d <- nbinomLRT(d, full= ~condition, reduced= ~1)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
sum(q.value < param_FDR)
sum(p.adjust(p.value, method="BH") < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotMA(d)
dev.off()
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
sum(q.value < 0.80)
sum(p.adjust(p.value, method="BH") < 0.05)
sum(p.adjust(p.value, method="BH") < 0.10)
sum(p.adjust(p.value, method="BH") < 0.30)
sum(p.adjust(p.value, method="BH") < 0.50)
sum(p.adjust(p.value, method="BH") < 0.70)
sum(p.adjust(p.value, method="BH") < 0.80)
1.と基本的に同じですが、TCCパッケージ中の記述方式です。
calcNormFactors関数実行部分で、iteration=0にしています。
これはTCCの長所である頑健な正規化パイプラインの機能をオフにすることに相当し、結果的にデフォルトのDESeq2パイプラインが走るようにしています。
in_f <- "data_hypodata_1vs1.txt"
out_f1 <- "hoge2.txt"
out_f2 <- "hoge2.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=0, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
sum(p.adjust(tcc$stat$p.value, method="BH") < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(tcc, FDR=param_FDR, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR=", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-result$rank))
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.30)
sum(tcc$stat$q.value < 0.50)
sum(tcc$stat$q.value < 0.70)
sum(tcc$stat$q.value < 0.80)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.05)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.10)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.30)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.50)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.70)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.80)
52,580 genes×2 samplesのカウントデータ(G1群1サンプル vs. G2群1サンプル)です。
DESeq2パッケージ中の記述方式です
plotMAのところでエラーが出てM-A plotは描画できません(DESeq2 ver. 1.12.0で2016年5月22日に確認)。
in_f <- "maqc_pooledreps_count_table.txt"
out_f1 <- "hoge3.txt"
out_f2 <- "hoge3.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(DESeq2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
d <- DESeq(d)
#d <- estimateSizeFactors(d)
##sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))
#d <- estimateDispersions(d)
#d <- nbinomLRT(d, full= ~condition, reduced= ~1)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
sum(q.value < param_FDR)
sum(p.adjust(p.value, method="BH") < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotMA(d)
dev.off()
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
sum(q.value < 0.80)
sum(p.adjust(p.value, method="BH") < 0.05)
sum(p.adjust(p.value, method="BH") < 0.10)
sum(p.adjust(p.value, method="BH") < 0.30)
sum(p.adjust(p.value, method="BH") < 0.50)
sum(p.adjust(p.value, method="BH") < 0.70)
sum(p.adjust(p.value, method="BH") < 0.80)
3.と基本的に同じですが、発現変動順にソートしています。
plotMAのところでエラーが出てM-A plotは描画できません(DESeq2 ver. 1.12.0で2016年5月22日に確認)。
in_f <- "maqc_pooledreps_count_table.txt"
out_f1 <- "hoge4.txt"
out_f2 <- "hoge4.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(DESeq2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
d <- DESeq(d)
#d <- estimateSizeFactors(d)
##sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))
#d <- estimateDispersions(d)
#d <- nbinomLRT(d, full= ~condition, reduced= ~1)
normalized <- counts(d, normalized=T)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
sum(q.value < param_FDR)
sum(p.adjust(p.value, method="BH") < param_FDR)
tmp <- cbind(rownames(data), normalized, p.value, q.value, ranking)
tmp <- tmp[order(ranking),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotMA(d)
dev.off()
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
sum(q.value < 0.80)
sum(p.adjust(p.value, method="BH") < 0.05)
sum(p.adjust(p.value, method="BH") < 0.10)
sum(p.adjust(p.value, method="BH") < 0.30)
sum(p.adjust(p.value, method="BH") < 0.50)
sum(p.adjust(p.value, method="BH") < 0.70)
sum(p.adjust(p.value, method="BH") < 0.80)
4.と基本的に同じですが、TCCパッケージ中の記述方式です。
calcNormFactors関数実行部分で、iteration=0にしています。
これはTCCの長所である頑健な正規化パイプラインの機能をオフにすることに相当し、結果的にデフォルトのDESeq2パイプラインが走るようにしています。
正規化後のデータ、発現変動順にソートして出力しています。M-A plotのところも変更しています。
in_f <- "maqc_pooledreps_count_table.txt"
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=0, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(tcc, FDR=param_FDR, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.30)
sum(tcc$stat$q.value < 0.50)
sum(tcc$stat$q.value < 0.70)
sum(tcc$stat$q.value < 0.80)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.05)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.10)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.30)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.50)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.70)
sum(p.adjust(tcc$stat$p.value, method="BH") < 0.80)
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC (Sun_2013)
TCCを用いたやり方を示します。
2016年5月21日に、TCC原著論文(Sun et al., BMC Bioinformatics, 2013)発表時の推奨解析パイプラインである、
iDEGES/DESeq-DESeqから、iDEGES/DESeq2-DESeq2に切り替えていましたが、2019年7月11日に元に戻しました。理由はDESeq2を使うとエラーが出るようになったからです。その後DESeqもBioconductorから削除されたので、2021年6月1日に内部的に利用する関数をedgeRパッケージのものに切り替えました。さらにその後、このスクリプトも動かなくなっていることを2022年4月28日に確認しました。現状それ以外の手段はありませんので、実行不可ということになります(辛川 涼眸 氏提供情報)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
head(result, n=3)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画したり、
DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
FDR閾値を満たす遺伝子数は0個、AUC = 0.798375であることが分かります。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge2.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_DEG <- 1:2000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
head(result, n=3)
sum(tcc$stat$q.value < param_FDR)
ranking <- rank(tcc$stat$p.value)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc, FDR=param_FDR)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
3. サンプルデータ8の26,221 genes×6 samplesのリアルデータ(data_arab.txt; mock 3サンプル vs. hrcc 3サンプル)の場合:
mock群の1番目(mock1)と3番目(mock3)のサブセットを抽出して複製なしデータとして取り扱っています。
「FDR閾値を満たすもの」と「fold-change閾値を満たすもの」それぞれのM-A plotを作成しています。
in_f <- "data_arab.txt"
out_f1 <- "hoge3.txt"
out_f2 <- "hoge3_FDR.png"
out_f3 <- "hoge3_FC.png"
param_subset <- c(1, 3)
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_FC <- 2
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <-data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
head(result, n=3)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
param_FC <- 2
M <- getResult(tcc)$m.value
hoge <- rep(1, length(M))
hoge[abs(M) > log2(param_FC)] <- 2
cols <- c("black", "magenta")
png(out_f3, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, col=cols, col.tag=hoge, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(", param_FC, "-fold)", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(abs(M) > log2(16))
sum(abs(M) > log2(8))
sum(abs(M) > log2(4))
sum(abs(M) > log2(2))
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
1.と基本的に同じで、出力のテキストファイルが正規化前のデータではなく正規化後のデータになっていて、発現変動順にソートしたものになっています。
in_f <- "data_hypodata_1vs1.txt"
out_f1 <- "hoge4.txt"
out_f2 <- "hoge4.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
52,580 genes×2 samplesのカウントデータ(G1群1サンプル vs. G2群1サンプル)です。
例題4と基本的に同じで、正規化後のデータ、発現変動順にソートして出力しています。M-A plotのところも変更しています。
in_f <- "maqc_pooledreps_count_table.txt"
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(tcc, FDR=param_FDR, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
ヒトHomo sapiens; HS)のメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3),
チンパンジー(Pan troglodytes; PT)のメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザル(Rhesus macaque; RM)のメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
つまり、以下のような感じです。FはFemale(メス)、MはMale(オス)を表します。
ヒト(1-6列目): HSF1, HSF2, HSF3, HSM1, HSM2, and HSM3
チンパンジー(7-12列目): PTF1, PTF2, PTF3, PTM1, PTM2, and PTM3
アカゲザル(13-18列目): RMF1, RMF2, RMF3, RMM1, RMM2, and RMM3
ここでは、ヒト1サンプル(G1群:HSF1) vs. アカゲザル1サンプル(G2群:RMF1)の比較を行っています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge6.txt"
out_f2 <- "hoge6.png"
param_subset <- c(1, 13)
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.10
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
例題6と基本的に同じで、ヒト1サンプル(G1群:HSF1) vs. チンパンジー1サンプル(G2群:PTF1)の比較を行っています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7.png"
param_subset <- c(1, 7)
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.10
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
例題6と基本的に同じで、ヒト1サンプル(G1群:HSF1) vs. ヒト1サンプル(G2群:HSM1)の比較を行っています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8.png"
param_subset <- c(1, 4)
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.10
param_fig <- c(430, 350)
param_mar <- c(4, 4, 0, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
dim(data)
head(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(tcc, FDR=param_FDR, xlim=c(-2, 17), ylim=c(-10, 11.5),
cex=0.8, cex.lab=1.2,
cex.axis=1.2, main="",
xlab="A = (log2(G2) + log2(G1))/2",
ylab="M = log2(G2) - log2(G1)")
legend("topright", c(paste("DEG(FDR =", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20, cex=1.2)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
- 多群間比較用の推奨ガイドライン提唱論文:Tang et al., BMC Bioinformatics, 2015
- DESeq2:Love et al., Genome Biol., 2014
- TCC:Sun et al., BMC Bioinformatics, 2013
- TbT正規化法(TCCに実装されたDEGESアルゴリズム提唱論文):Kadota et al., Algorithms Mol. Biol., 2012
- DESeq:Anders and Huber, Genome Biol, 2010
- Blekhman et al., Genome Res., 2010
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | DESeq (Anders_2010)
Anders and Huberの(AH)正規化(Anders_2010)を実行したのち、
DESeqパッケージ中のnegative binomial testで発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行う
DESeqパッケージ内のオリジナルの手順を示します。
このパッケージはもはや存在しないので動きません(2021.06.01追加)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
library(DESeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
cds <- newCountDataSet(data, data.cl)
cds <- estimateSizeFactors(cds)
sizeFactors(cds)
cds <- estimateDispersions(cds, method="blind", sharingMode="fit-only")
out <- nbinomTest(cds, 1, 2)
p.value <- out$pval
p.value[is.na(p.value)] <- 1
q.value <- out$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
logratio <- out$log2FoldChange
head(out, n=3)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking, logratio)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
1.と基本的に同じで、param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画したり、
このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
AUC = 0.7811263ですね。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge2.txt"
param_G1 <- 1
param_G2 <- 1
param_DEG <- 1:2000
param_FDR <- 0.1
library(DESeq)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
cds <- newCountDataSet(data, data.cl)
cds <- estimateSizeFactors(cds)
sizeFactors(cds)
cds <- estimateDispersions(cds, method="blind", sharingMode="fit-only")
out <- nbinomTest(cds, 1, 2)
p.value <- out$pval
p.value[is.na(p.value)] <- 1
q.value <- out$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
logratio <- out$log2FoldChange
head(out, n=3)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking, logratio)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plotMA(out)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
TCCパッケージを用いて同様の解析を行うやり方です。
出力ファイルのa.value列がlogratioに相当し、q.value列上でFDR閾値を決めます。(内部的な細かい話ですが...)estimateDispersions関数実行時に、fitType="parametric"を最初に試して、エラーが出たら自動的に"local"に変更しています。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge3.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.10
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc,iteration=0)
tcc <- estimateDE(tcc, FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
head(result, n=3)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
TCCパッケージを用いて同様の解析を行うやり方です。
出力ファイルのa.value列がlogratioに相当し、q.value列上でFDR閾値を決めます。(内部的な細かい話ですが...)estimateDispersions関数実行時に、fitType="parametric"を最初に試して、
エラーが出たら自動的に"local"に変更しています。param_FDRで指定した閾値を満たすDEGをマゼンタ色にして描画したり、
このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge4.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.10
param_DEG <- 1:2000
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc,iteration=0)
tcc <- estimateDE(tcc, FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
head(result, n=3)
sum(tcc$stat$q.value < param_FDR)
ranking <- tcc$stat$rank
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
plot(tcc, FDR=param_FDR)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 2群間 | 対応なし | 複製なし | edgeR(Robinson_2010)
edgeRパッケージを用いて発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。estimateGLMCommonDisp関数実行部分のオプションは、
edgeRUsersGuide.pdfの
反復なしの場合(2.11 What to do if you have no replicates)の、選択肢3に相当します。
verbose=TRUEは、計算したBCV値を表示するオプションです。common.dispersion = 0.45507、BCV = 0.6746、AUC = 0.771165ですね。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
d <- DGEList(counts=data,group=data.cl)
d <- calcNormFactors(d)
d <- estimateGLMCommonDisp(d, method="deviance",
robust=TRUE, subset=NULL, verbose=TRUE)
out <- exactTest(d)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
例題1とほとんど同じで、estimateGLMCommonDisp関数実行部分のオプションが若干異なります。
common.dispersion = 0.4310842、BCV = 0.6566、AUC = 0.7726519ですね。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge2.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
d <- DGEList(counts=data,group=data.cl)
d <- calcNormFactors(d)
d <- estimateGLMCommonDisp(d, verbose=TRUE)
out <- exactTest(d)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
non-DEGの正しい情報を与えることで、common.dispersion値が正しく小さい値になるかどうかを試すコードです。
estimateDisp関数実行部分のオプションは、
edgeRUsersGuide.pdfの
反復なしの場合(2.11 What to do if you have no replicates)の、選択肢4に相当します。
common.dispersion = 0.2544829、BCV = 0.504463、AUC = 0.7784515ですね。
non-DEGの正しい情報を与えているのでうまくいって当然ではありますが、
反復なしデータでもFDR0.05を満たすのが53個得られていることがわかります。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge3.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
d <- DGEList(counts=data,group=data.cl)
d <- calcNormFactors(d)
d1 <- d
d1$samples$group <- 1
d1 <- estimateDisp(d1[2001:10000, ],
trend="none", tagwise=FALSE, verbose=TRUE)
d$common.dispersion <- d1$common.dispersion
d$common.dispersion
sqrt(d$common.dispersion)
out <- exactTest(d)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
例題3と基本的に同じですが、estimateDisp関数ではなく、estimateGLMCommonDisp関数を使っています。
common.dispersion = 0.24691、BCV = 0.4969、AUC = 0.7783244ですね。
例題3と同じく、反復なしデータでもFDR0.05を満たすのが64個得られていることがわかります。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge4.txt"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- as.matrix(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
d <- DGEList(counts=data,group=data.cl)
d <- calcNormFactors(d)
d1 <- estimateGLMCommonDisp(d[2001:10000, ], verbose=TRUE)
d$common.dispersion <- d1$common.dispersion
out <- exactTest(d)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
param_DEG <- 1:2000
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- an exact test for negative binomial distribution:Robinson and Smyth, Biostatistics, 2008
- Robinson and Smyth, Bioinformatics, 2007
- McCarthy et al., Nucleic Acids Res., 2012
解析 | 発現変動 | 2群間 | 対応あり | について
実験デザインが以下のような場合にこのカテゴリーに属す方法を適用します:
Aさんの正常サンプル(or "time_point1" or "Group1")
Bさんの正常サンプル(or "time_point1" or "Group1")
Cさんの正常サンプル(or "time_point1" or "Group1")
Aさんの腫瘍サンプル(or "time_point2" or "Group2")
Bさんの腫瘍サンプル(or "time_point2" or "Group2")
Cさんの腫瘍サンプル(or "time_point2" or "Group2")
2015年11月に調査した結果をリストアップします。
R用:
- edgeR:Robinson et al., Bioinformatics, 2010
- DESeq:Anders and Huber, Genome Biol., 2010
- baySeq:Hardcastle and Kelly, BMC Bioinformatics, 2013
- TCC:Sun et al., BMC Bioinformatics, 2013
- DESeq2:Love et al., Genome Biol., 2014
解析 | 発現変動 | 2群間 | 対応あり | 複製なし | TCC中のDEGES/edgeR-edgeR (Sun_2013)
DEGES/edgeR正規化(Sun et al., 2013)を実行したのち、
edgeRパッケージ中のGLM (一般化線形モデル; Generalized Linear Model)に基づく方法
(McCarthy et al., 2012)で発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
TCC原著論文中のDEGES/edgeR-edgeRという解析パイプラインに相当します。
Bioconductor ver. 2.13で利用可能なTCC ver. 1.2.0ではまだ実装されていないので、
ここでは、edgeRパッケージ中の関数のみを用いて対応のあるサンプル(paired samples)データ解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
d <- DGEList(counts=data)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ pair + cl)
FDR <- 0.1
floorPDEG <- 0.05
### STEP 1 ###
d <- calcNormFactors(d)
d$samples$norm.factors
### STEP 2 ###
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
if (sum(q.value < FDR) > (floorPDEG * nrow(data))){
is.DEG <- as.logical(q.value < FDR)
} else {
is.DEG <- as.logical(rank(p.value, ties.method="min") <= nrow(data)*floorPDEG)
}
### STEP 3 ###
d <- DGEList(counts=data[!is.DEG, ])
d <- calcNormFactors(d)
norm.factors <- d$samples$norm.factors*colSums(data[!is.DEG, ])/colSums(data)
norm.factors <- norm.factors/mean(norm.factors)
norm.factors
d <- DGEList(counts=data)
d$samples$norm.factors <- norm.factors
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
1.と全く同じ結果が得られますが、デザイン行列designの作成のところで順番を入れ替えています。
それに伴い、検定時のglmLRT関数実行時に、coefで指定する列情報を2列目に変更しています。1.ではデフォルトがdesign行列の最後の列なので、何も指定しなくていいのです。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
d <- DGEList(counts=data)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ cl + pair)
FDR <- 0.1
floorPDEG <- 0.05
### STEP 1 ###
d <- calcNormFactors(d)
d$samples$norm.factors
### STEP 2 ###
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
if (sum(q.value < FDR) > (floorPDEG * nrow(data))){
is.DEG <- as.logical(q.value < FDR)
} else {
is.DEG <- as.logical(rank(p.value, ties.method="min") <= nrow(data)*floorPDEG)
}
### STEP 3 ###
d <- DGEList(counts=data[!is.DEG, ])
d <- calcNormFactors(d)
norm.factors <- d$samples$norm.factors*colSums(data[!is.DEG, ])/colSums(data)
norm.factors <- norm.factors/mean(norm.factors)
norm.factors
d <- DGEList(counts=data)
d$samples$norm.factors <- norm.factors
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit, coef=2)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
DEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_DEG <- 1:2000
library(edgeR)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
d <- DGEList(counts=data)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ pair + cl)
FDR <- 0.1
floorPDEG <- 0.05
### STEP 1 ###
d <- calcNormFactors(d)
d$samples$norm.factors
### STEP 2 ###
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
if (sum(q.value < FDR) > (floorPDEG * nrow(data))){
is.DEG <- as.logical(q.value < FDR)
} else {
is.DEG <- as.logical(rank(p.value, ties.method="min") <= nrow(data)*floorPDEG)
}
### STEP 3 ###
d <- DGEList(counts=data[!is.DEG, ])
d <- calcNormFactors(d)
norm.factors <- d$samples$norm.factors*colSums(data[!is.DEG, ])/colSums(data)
norm.factors <- norm.factors/mean(norm.factors)
norm.factors
d <- DGEList(counts=data)
d$samples$norm.factors <- norm.factors
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
4. TCCパッケージ中の1,000 genes×6 samplesのカウントデータ(hypoData)の場合:
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_200までがDEG (最初の180個がG1群で高発現、残りの20個がG2群で高発現)
gene_201〜gene_1000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
out_f <- "hoge4.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
library(TCC)
data(hypoData)
data <- hypoData
d <- DGEList(counts=data)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ pair + cl)
FDR <- 0.1
floorPDEG <- 0.05
### STEP 1 ###
d <- calcNormFactors(d)
d$samples$norm.factors
### STEP 2 ###
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
if (sum(q.value < FDR) > (floorPDEG * nrow(data))){
is.DEG <- as.logical(q.value < FDR)
} else {
is.DEG <- as.logical(rank(p.value, ties.method="min") <= nrow(data)*floorPDEG)
}
### STEP 3 ###
d <- DGEList(counts=data[!is.DEG, ])
d <- calcNormFactors(d)
norm.factors <- d$samples$norm.factors*colSums(data[!is.DEG, ])/colSums(data)
norm.factors <- norm.factors/mean(norm.factors)
norm.factors
d <- DGEList(counts=data)
d$samples$norm.factors <- norm.factors
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. TCCパッケージ中の1,000 genes×6 samplesのカウントデータ(hypoData)の場合:
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_200までがDEG (最初の180個がG1群で高発現、残りの20個がG2群で高発現)
gene_201〜gene_1000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
4.と全く同じ結果が得られますが、デザイン行列designの作成のところで順番を入れ替えています。
それに伴い、検定時のglmLRT関数実行時に、coefで指定する列情報を2列目に変更しています。1.ではデフォルトがdesign行列の最後の列なので、何も指定しなくていいのです。
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
library(TCC)
data(hypoData)
data <- hypoData
d <- DGEList(counts=data)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ cl + pair)
FDR <- 0.1
floorPDEG <- 0.05
### STEP 1 ###
d <- calcNormFactors(d)
d$samples$norm.factors
### STEP 2 ###
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit, coef=2)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
if (sum(q.value < FDR) > (floorPDEG * nrow(data))){
is.DEG <- as.logical(q.value < FDR)
} else {
is.DEG <- as.logical(rank(p.value, ties.method="min") <= nrow(data)*floorPDEG)
}
### STEP 3 ###
d <- DGEList(counts=data[!is.DEG, ])
d <- calcNormFactors(d)
norm.factors <- d$samples$norm.factors*colSums(data[!is.DEG, ])/colSums(data)
norm.factors <- norm.factors/mean(norm.factors)
norm.factors
d <- DGEList(counts=data)
d$samples$norm.factors <- norm.factors
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit, coef=2)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 発現変動 | 2群間 | 対応あり | 複製なし | TCC中のDEGES/DESeq-DESeq (Sun_2013)
書きかけです。
DEGES/DESeq正規化(Sun et al., 2013)を実行したのち、
DESeqパッケージ中のGLM (一般化線形モデル; Generalized Linear Model)に基づく方法
で発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
TCC原著論文中のDEGES/DESeq-DESeqという解析パイプラインに相当します。
Bioconductor ver. 2.13で利用可能なTCC ver. 1.2.0ではまだ実装されていないので、
ここでは、DESeqパッケージ中の関数のみを用いて対応のあるサンプル(paired samples)データ解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
TCC ver. 1.3.2で2014年4月に利用可能予定のコードです。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(DESeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
d <- DGEList(counts=data)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ pair + cl)
FDR <- 0.1
floorPDEG <- 0.05
### STEP 1 ###
d <- calcNormFactors(d)
d$samples$norm.factors
### STEP 2 ###
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
if (sum(q.value < FDR) > (floorPDEG * nrow(data))){
is.DEG <- as.logical(q.value < FDR)
} else {
is.DEG <- as.logical(rank(p.value, ties.method="min") <= nrow(data)*floorPDEG)
}
### STEP 3 ###
d <- DGEList(counts=data[!is.DEG, ])
d <- calcNormFactors(d)
norm.factors <- d$samples$norm.factors*colSums(data[!is.DEG, ])/colSums(data)
norm.factors <- norm.factors/mean(norm.factors)
norm.factors
d <- DGEList(counts=data)
d$samples$norm.factors <- norm.factors
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 発現変動 | 2群間 | 対応あり | 複製なし | DESeq (Anders_2010)
DESeqパッケージを用いて対応のあるサンプル(paired samples)データ解析を行うやり方を示します。
TCCパッケージでも内部的にDESeqを呼び出して実行可能なので、そのやり方も示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
TCC ver. 1.3.2で2014年4月に利用可能予定のコードです。getResult関数のところでエラーが出るようですね。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
design.cl <- data.frame(
group=c(rep(1, param_G1), rep(2, param_G2)),
pair=c(1:param_G1, 1:param_G2)
)
tcc <- new("TCC", data, design.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0, paired=T)
tcc$norm.factors
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR, paired=T)
#result <- getResult(tcc, sort=FALSE)
#sum(tcc$stat$q.value < param_FDR)
p.value <- tcc$stat$p.value
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
#tmp <- cbind(rownames(tcc$count), tcc$count, result)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
DEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
TCC ver. 1.3.2で2014年4月に利用可能予定のコードです。getResult関数のところでエラーが出るようですね。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_DEG <- 1:2000
library(TCC)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
design.cl <- data.frame(
group=c(rep(1, param_G1), rep(2, param_G2)),
pair=c(1:param_G1, 1:param_G2)
)
tcc <- new("TCC", data, design.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0, paired=T)
tcc$norm.factors
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR, paired=T)
#result <- getResult(tcc, sort=FALSE)
#sum(tcc$stat$q.value < param_FDR)
p.value <- tcc$stat$p.value
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
#tmp <- cbind(rownames(tcc$count), tcc$count, result)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
1.と同じ入力形式で、TCCパッケージを使わずにDESeqパッケージ中の関数のみで行うやり方です。TCCのほうが手順的にも簡便であることがわかります。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(DESeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
pair <- c(1:param_G1, 1:param_G2)
design.cl <- data.frame(
group=as.factor(data.cl),
pair=as.factor(pair)
)
cds <- newCountDataSet(data, design.cl)
cds <- estimateSizeFactors(cds)
sizeFactors(cds)
cds <- estimateDispersions(cds, method="blind", sharingMode="fit-only")
fit1 <- fitNbinomGLMs(cds, count ~ as.factor(data.cl) + as.factor(pair))
fit0 <- fitNbinomGLMs(cds, count ~ as.factor(pair))
p.value <- nbinomGLMTest(fit1, fit0)
p.value[is.na(p.value)] <- 1
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
4. TCCパッケージ中の1,000 genes×6 samplesのカウントデータ(hypoData)の場合:
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_200までがDEG (最初の180個がG1群で高発現、残りの20個がG2群で高発現)
gene_201〜gene_1000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
TCC ver. 1.3.2で2014年4月に利用可能予定のコードです。getResult関数のところでエラーが出るようですね。
out_f <- "hoge4.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(TCC)
library(DESeq)
data(hypoData)
data <- hypoData
design.cl <- data.frame(
group=c(rep(1, param_G1), rep(2, param_G2)),
pair=c(1:param_G1, 1:param_G2)
)
tcc <- new("TCC", data, design.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0, paired=T)
tcc$norm.factors
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR, paired=T)
#result <- getResult(tcc, sort=FALSE)
#sum(tcc$stat$q.value < param_FDR)
p.value <- tcc$stat$p.value
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
#tmp <- cbind(rownames(tcc$count), tcc$count, result)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5. TCCパッケージ中の1,000 genes×6 samplesのカウントデータ(hypoData)の場合:
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_200までがDEG (最初の180個がG1群で高発現、残りの20個がG2群で高発現)
gene_201〜gene_1000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
DEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
TCC ver. 1.3.2で2014年4月に利用可能予定のコードです。getResult関数のところでエラーが出るようですね。
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_DEG <- 1:200
library(TCC)
library(ROC)
library(DESeq)
data(hypoData)
data <- hypoData
design.cl <- data.frame(
group=c(rep(1, param_G1), rep(2, param_G2)),
pair=c(1:param_G1, 1:param_G2)
)
tcc <- new("TCC", data, design.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0, paired=T)
tcc$norm.factors
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR, paired=T)
#result <- getResult(tcc, sort=FALSE)
#sum(tcc$stat$q.value < param_FDR)
p.value <- tcc$stat$p.value
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
#tmp <- cbind(rownames(tcc$count), tcc$count, result)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
6. TCCパッケージ中の1,000 genes×6 samplesのカウントデータ(hypoData)の場合:
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_200までがDEG (最初の180個がG1群で高発現、残りの20個がG2群で高発現)
gene_201〜gene_1000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
4.と同じ入力形式で、TCCパッケージを使わずにDESeqパッケージ中の関数のみで行うやり方です。TCCのほうが手順的にも簡便であることがわかります。
out_f <- "hoge6.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(DESeq)
library(TCC)
data(hypoData)
data <- hypoData
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
pair <- c(1:param_G1, 1:param_G2)
design.cl <- data.frame(
group=as.factor(data.cl),
pair=as.factor(pair)
)
cds <- newCountDataSet(data, design.cl)
cds <- estimateSizeFactors(cds)
sizeFactors(cds)
cds <- estimateDispersions(cds, method="blind", sharingMode="fit-only")
fit1 <- fitNbinomGLMs(cds, count ~ as.factor(data.cl) + as.factor(pair))
fit0 <- fitNbinomGLMs(cds, count ~ as.factor(pair))
p.value <- nbinomGLMTest(fit1, fit0)
p.value[is.na(p.value)] <- 1
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 発現変動 | 2群間 | 対応あり | 複製なし | edgeR(Robinson_2010)
edgeRパッケージを用いて対応のあるサンプル(paired samples)データ解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
d <- DGEList(data)
d <- calcNormFactors(d)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ pair + cl)
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
Biological replicatesを模倣したシミュレーションデータ(G1群3サンプル vs. G2群3サンプル)です。
gene_1〜gene_2000までがDEG (最初の1800個がG1群で高発現、残りの200個がG2群で高発現)
gene_2001〜gene_10000までがnon-DEGであることが既知です。
G1群の3つのサンプル(G1_1, G1_2, G1_3)とG2群の3つのサンプル(G2_1, G2_2, G2_3)が対応しているという仮定で解析を行います。
例えば、G1_1とG2_1がAさん、G1_2とG2_2がBさん、そしてG1_3とG2_3がCさんというイメージです。したがって、G1群とG2群のサンプル数が異なることはアリエマセン。
DEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算を行う例です。
in_f <- "data_hypodata_3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_DEG <- 1:2000
library(edgeR)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
d <- DGEList(data)
d <- calcNormFactors(d)
pair <- as.factor(c(1:param_G1, 1:param_G2))
cl <- as.factor(c(rep(1, param_G1), rep(2, param_G2)))
design <- model.matrix(~ pair + cl)
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
out <- glmLRT(fit)
#tmp <- topTags(out, n=nrow(data), sort.by="none")
p.value <- out$table$PValue
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 3群間 | 対応なし | について
実験デザインが以下のような場合にこのカテゴリーに属す方法を適用します:
Aさんの肝臓サンプル(or "time_point1" or "Group1")
Bさんの肝臓サンプル(or "time_point1" or "Group1")
Cさんの肝臓サンプル(or "time_point1" or "Group1")
Dさんの腎臓サンプル(or "time_point2" or "Group2")
Eさんの腎臓サンプル(or "time_point2" or "Group2")
Fさんの腎臓サンプル(or "time_point2" or "Group2")
Gさんの大腸サンプル(or "time_point3" or "Group3")
Hさんの大腸サンプル(or "time_point3" or "Group3")
Iさんの大腸サンプル(or "time_point3" or "Group3")
基礎では、ANOVAのような「どこかの群間で発現に差がある遺伝子の検出」のやり方を示します。
応用では、「指定した群間で発現に差がある遺伝子の検出」のやり方を示します。
GLM LRT法などが有名です。GLMは一般化線形モデル(generalized linear model)の略、そしてLRTは尤度比検定(log-likelihood ratio test)の略です。
基本的には、「全ての群の情報を含むfull model」と「特定の情報を除くreduced model」の2つモデルを作成し、その二つのモデルの尤度に差があるかどうかを検定するやり方です。
designというオブジェクトがfull modelに相当します。基本的には、デザイン行列中のparam_coefで指定した列を除くことでreduced modelを作成してLRTを行っています。
EBSeqやbaySeqは、考えられる様々なパターンのうちどのパターンへの当てはまりが最も良いかというposterior probabilityを出力します。
手法比較論文(Tang et al., BMC Bioinformatics, 2015)
中では、複製ありデータ用はTCCのDEGESパイプライン内部でedgeRを用いたEEE-E、
そして複製なしデータ用はDEGESパイプライン内部でDESeq2を用いたSSS-Sを多群間比較用の推奨ガイドラインとして結論づけています。
EEE-EはTCC論文中のiDEGES/edgeR-edgeRパイプラインと、そしてSSS-SはiDEGES/DESeq2-DESeq2パイプラインに相当します。
2016年10月に調査した結果をリストアップします。
R用:
- edgeR:Robinson et al., Bioinformatics, 2010
- baySeq:Hardcastle and Kelly, BMC Bioinformatics, 2010
- DESeq:Anders and Huber, Genome Biol, 2010
- PoissonSeq:Li et al., Biostatistics, 2012
- SAMseq:Li and Tibshirani, Stat Methods Med Res., 2013
- EBSeq:Leng et al., Bioinformatics, 2013
- TCC:Sun et al., BMC Bioinformatics, 2013
- DESeq2:Love et al., Genome Biol., 2014
- deGPS:Chu et al., BMC Genomics, 2015
- multiDE:Kang et al., BMC Bioinformatics, 2016
- derfinder:Collado-Torres et al., Nucleic Acids Res., 2016
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | DESeq2(Love_2014)
DESeq2パッケージ (Love et al., Genome Biol., 2014)を用いるやり方を示します。
DESeq2はTCCパッケージ (Sun et al., BMC Bioinformatics, 2013)内で用いることができるので、そのやり方を中心に述べます。
デフォルトのDESeq2の手順に相当します。
ここでやっていることはANOVAのような「どこかの群間で発現に差がある遺伝子を検出」です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。TCCパッケージを用いるやり方です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. サンプルデータ15の10,000 genes×9 samplesのシミュレーションデータ(data_hypodata_3vs3vs3.txt)と同じものをTCCパッケージ内部で作成する場合:
1.と同じ結果が得られます。
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="deseq2", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3.と同じ結果が得られます。
out_f <- "hoge4.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="deseq2", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
1.と基本的に同じで、出力のテキストファイルが正規化前のデータではなく正規化後のデータになっていて、発現変動順にソートしたものになっています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", iteration=0)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
1.と基本的に同じですが、full modelとreduced modelを情報を明示的に指定しています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge6.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", iteration=0)
tcc$group <- data.frame(condition = as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR,
full= ~condition, reduced= ~1)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3.と基本的に同じで、TCCパッケージを使わずにDESeq2パッケージ中の関数のみで行うやり方です。
TCCのほうが手順的にも簡便であることがわかります。
指定したFDR閾値を満たす遺伝子数が異なるのは、q-value算出方法の違いに起因します。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge7.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(DESeq2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
d <- estimateSizeFactors(d)
sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))
d <- estimateDispersions(d)
d <- nbinomLRT(d, full= ~condition, reduced= ~1)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
sum(q.value < param_FDR)
sum(p.adjust(p.value, method="BH") < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_2000がG2群で5倍高発現、gene_2001〜gene_3000がG3群で5倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算も行っています。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge8.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(TCC)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
ranking <- rank(tcc$stat$p.value)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
8.と基本的に同じですが、TCCパッケージではなくDESeq2パッケージ中の関数を利用しています。
指定したFDR閾値を満たす遺伝子数が異なるのは、q-value算出方法の違いに起因します。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge9.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(DESeq2)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
d <- estimateSizeFactors(d)
sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))
d <- estimateDispersions(d)
d <- nbinomLRT(d, full= ~condition, reduced= ~1)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
sum(q.value < param_FDR)
sum(p.adjust(p.value, method="BH") < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | TCC(Sun_2013)
TCCを用いたやり方を示します。
内部的にiDEGES/edgeR(Sun_2013)正規化を実行したのち、
edgeRパッケージ中のGLM LRT法で発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行っています。
TCC原著論文中のiDEGES/edgeR-edgeRという解析パイプラインに相当します。
TCC原著論文(Sun et al., BMC Bioinformatics, 2013)では
3群間複製ありデータ用の推奨パイプラインを示していませんでしたが、多群間比較用の推奨ガイドライン提唱論文
(Tang et al., BMC Bioinformatics, 2015)
で推奨しているパイプライン"EEE-E"が"iDEGES/edgeR-edgeR"と同じものです。この2つの論文を引用し、安心してご利用ください。
尚、ここでやっていることはANOVAのような「どこかの群間で発現に差がある遺伝子を検出」です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
1.と同じ結果が得られます。
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3.と同じ結果が得られます。
out_f <- "hoge4.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
1.と基本的に同じで、出力のテキストファイルが正規化前のデータではなく正規化後のデータになっていて、発現変動順にソートしたものになっています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
1.と基本的に同じですが、デザイン行列designと係数coefを明示的に作成および指定しています。
coefに入っている数値は、length(unique(data.cl))の値が3になるので、2:3、つまり2と3になります。
これがANOVA的などこかの群間で発現変動しているものを検出するときに指定する値です。
感覚的にはdesign[,2] - design[,3]をやっていることになり、「G1で-1, G2で0, G3で1」と全ての群で異なる数値が割り振られることになります。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge6.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
design <- model.matrix(~ as.factor(data.cl))
coef <- 2:length(unique(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=coef)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
5.と基本的に同じで、入力ファイルが違うだけです。Blekhman et al., Genome Res., 2010の
20,689 genes×18 samplesのカウントデータです。ヒト(HS)、チンパンジー(PT)、アカゲザル(RM)の3生物種間比較です。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge7.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
例題7と基本的に同じで、各生物種につき最初の5サンプル分を抽出して5反復の3群間比較を行っています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge8.txt"
param_subset <- c(1:5, 7:11, 13:17)
param_G1 <- 5
param_G2 <- 5
param_G3 <- 5
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
例題7と基本的に同じで、各生物種につき最初の4サンプル分を抽出して4反復の3群間比較を行っています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge9.txt"
param_subset <- c(1:4, 7:10, 13:16)
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
例題7と基本的に同じで、各生物種につき最初の3サンプル分を抽出して3反復の3群間比較を行っています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge10.txt"
param_subset <- c(1:3, 7:9, 13:15)
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
- 多群間比較用の推奨ガイドライン提唱論文:Tang et al., BMC Bioinformatics, 2015
- TCC:Sun et al., BMC Bioinformatics, 2013
- TbT正規化法(TCCに実装されたDEGESアルゴリズム提唱論文):Kadota et al., Algorithms Mol. Biol., 2012
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR中のGLM論文:McCarthy et al., Nucleic Acids Res., 2012
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | EBSeq(Leng_2013)
EBSeqパッケージ(Leng et al., 2013)を用いて
発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
ANOVAのような「どこかの群間で発現に差がある遺伝子を検出」ではなく、EBSeqで定義したいくつかの発現パターンのどれに相当するかまで示してくれるので、
「どこで発現に差があるのか」を調べるpost-hoc testを行う必要がないのが特徴です。
出力結果ファイルを眺めると
Pattern1, Pattern2, ..., Pattern5までの列のところに遺伝子ごとのPosterior Probability (PP)値が与えられています。
PP値が一番大きい(つまり1に近い)パターンに、その遺伝子が属するということになります:
Pattern1は(1, 1, 1)なのでnon-DEGの発現パターン
Pattern2は(1, 1, 2)なのでG3群で発現変動(高発現または低発現)
Pattern3は(1, 2, 1)なのでG2群で発現変動
Pattern4は(1, 2, 2)なのでG1群で発現変動
Pattern5は(1, 2, 3)なので、全ての群間で発現変動
一番右側のout$MAP列を見る方が手っ取り早いかもしれません。どの遺伝子がどのパターンに属するかが一目瞭然です。
全体的な発現変動の度合いによるANOVA-likeなランキングは、例題5などを参考にしてPattern1のPP値に基づいて行えばよいです。
Pattern1のPP値の低さはnon-DEGである確率の低さを意味するからです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_2000がG2群で5倍高発現、gene_2001〜gene_3000がG3群で5倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge1.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
size.factors <- MedianNorm(data)
size.factors
PosParti <- GetPatterns(data.cl)
Parti <- PosParti
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
1.と基本的に同じですが、調べるパターンの中からPattern5を排除するやり方です。
DegPack (An et al., 2014)
はまさにPattern5のような「どの群間でも同じパターンではない遺伝子」の検出を目的としていますが、
そういうものは興味の対象外という場合にここを利用します。尚、param_patternで指定している1:4は
c(1, 2, 3, 4)と同じ意味です。
1.でPattern5への当てはまりがよいとされていたgene_32が、Pattern4になっていることがわかります。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge2.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_pattern <- 1:4
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
size.factors <- MedianNorm(data)
size.factors
PosParti <- GetPatterns(data.cl)
Parti <- PosParti[param_pattern, ]
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
1.と基本的に同じですが、調べるパターンの中からPattern2, 3, 4を排除して、Pattern1と5のみを想定するやり方です。
DegPack (An et al., 2014)
と似た解析である「どの群間でも同じパターンではない遺伝子」の検出をやっていることに相当します。
私は通常の解析ではやりません。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge3.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_pattern <- c(1, 5)
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
size.factors <- MedianNorm(data)
size.factors
PosParti <- GetPatterns(data.cl)
Parti <- PosParti[param_pattern, ]
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
ここでは、ヒト(G1群)6サンプル, チンパンジー(G2群)6サンプル,
アカゲザル(G3群)6サンプルの3群間比較を行います。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge4.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
size.factors <- MedianNorm(data)
size.factors
PosParti <- GetPatterns(data.cl)
Parti <- PosParti
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。AUC値も表示させています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
size.factors <- MedianNorm(data)
size.factors
PosParti <- GetPatterns(data.cl)
Parti <- PosParti
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | SAMseq(Li_2013)
samrパッケージ中のSAMseq (Li and Tibshirani, 2013)を用いて
発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_2000がG2群で5倍高発現、gene_2001〜gene_3000がG3群で5倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。TCCパッケージでSAMseqを実行するやり方です。
このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算も行っています。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge1.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(TCC)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
set.seed(2015)
tcc <- estimateDE(tcc, test.method="samseq", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
ranking <- rank(tcc$stat$p.value)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
tcc$private$simulation <- TRUE
tcc$simulation$trueDEG <- obj
calcAUCValue(tcc)
シミュレーションデータ(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_2000がG2群で5倍高発現、gene_2001〜gene_3000がG3群で5倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。samrパッケージでSAMseqを実行する通常のやり方です。
このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算も行っています。同じAUC値(=0.8965379)が得られることが分かります。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge2.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(samr)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
set.seed(2015)
out <- SAMseq(data, data.cl, resp.type="Multiclass")
p.value <- samr.pvalues.from.perms(out$samr.obj$tt, out$samr.obj$ttstar)
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
シミュレーションデータ(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_2000がG2群で5倍高発現、gene_2001〜gene_3000がG3群で5倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。samrパッケージでSAMseqを実行する通常のやり方です。
このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算も行っています。1.や2.と若干異なるAUC値(=0.896619)が得られることが分かります。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge3.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(samr)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
set.seed(2015)
out <- SAMseq(data, data.cl, resp.type="Multiclass", fdr.output=1)
hoge <- rbind(out$siggenes.table$genes.up, out$siggenes.table$genes.lo)
q.value <- rep(1, nrow(data))
names(q.value) <- rownames(data)
q.value[as.numeric(hoge[, "Gene Name"])] <- as.numeric(hoge[, "q-value(%)"])/100
ranking <- rank(q.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
3.と基本的に同じで、記述形式をちょっと変更しているだけです。
SAMseq関数実行時の「nperms = 100, nresamp = 20」はデフォルトの数値を明示しているだけです。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge4.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(samr)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
set.seed(2015)
out <- SAMseq(data, data.cl, nperms = 100, nresamp = 20,
geneid = rownames(data), genenames = rownames(data),
resp.type="Multiclass", fdr.output=1)
hoge <- rbind(out$siggenes.table$genes.up, out$siggenes.table$genes.lo)
q.value <- rep(1, nrow(data))
names(q.value) <- rownames(data)
q.value[match(hoge[,1], rownames(data))] <- as.numeric(hoge[, "q-value(%)"])/100
ranking <- rank(q.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | DESeq(Anders_2010)
DESeqパッケージを用いて発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行うやり方(DESeq coupled with DESeq normalization; DESeq-DESeq)を示します。
デフォルトのDESeqの手順に相当します。TCCパッケージを用いても同様のことができますので、動作確認も兼ねて両方のやり方を示しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. サンプルデータ15の10,000 genes×9 samplesのシミュレーションデータ(data_hypodata_3vs3vs3.txt)と同じものをTCCパッケージ内部で作成する場合:
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
1.と同じ入力形式で、TCCパッケージを使わずにDESeqパッケージ中の関数のみで行うやり方です。TCCのほうが手順的にも簡便であることがわかります。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(DESeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
cds <- newCountDataSet(data, data.cl)
cds <- estimateSizeFactors(cds)
sizeFactors(cds)
cds <- estimateDispersions(cds, method="pooled")
fit1 <- fitNbinomGLMs(cds, count ~ as.factor(data.cl))
fit0 <- fitNbinomGLMs(cds, count ~ 1)
p.value <- nbinomGLMTest(fit1, fit0)
p.value[is.na(p.value)] <- 1
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge4.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
out_f <- "hoge5.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="deseq", iteration=0)
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
3.と同じ入力形式で、TCCパッケージを使わずにDESeqパッケージ中の関数のみで行うやり方です。TCCのほうが手順的にも簡便であることがわかります。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge6.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(DESeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
cds <- newCountDataSet(data, data.cl)
cds <- estimateSizeFactors(cds)
sizeFactors(cds)
cds <- estimateDispersions(cds, method="pooled")
fit1 <- fitNbinomGLMs(cds, count ~ as.factor(data.cl))
fit0 <- fitNbinomGLMs(cds, count ~ 1)
p.value <- nbinomGLMTest(fit1, fit0)
p.value[is.na(p.value)] <- 1
q.value <- p.adjust(p.value, method="BH")
ranking <- rank(p.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | baySeq(Hardcastle_2010)
baySeqを用いて発現変動遺伝子(Differentially Expressed Genes; DEGs)
検出を行うやり方を示します。
ANOVAのような「どこかの群間で発現に差がある遺伝子を検出」ではなく、baySeqで定義したいくつかの発現パターンのどれに相当するかまで示してくれるので、
「どこで発現に差があるのか」を調べるpost-hoc testを行う必要がないのが特徴です。
2018年7月9日に、ここの全ての例題にset.seed(2015)を埋め込みました。
試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(2015)の前に#を入れましょう。
つまり「set.seed(2015)」->「#set.seed(2015)」です。
2015はタネ番号なので3や496などでも構いません。
例題5以降がより適切だと思っておりますので、そちらをご利用ください(長部 高之 氏提供情報をベースに作成;2018.07.09)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。約3分。AUC値は0.875086ですね。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
param_samplesize <- 2000
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
NDE <- factor(c(rep("NDE", param_G1), rep("NDE", param_G2), rep("NDE", param_G3)))
DE <- factor(c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3)))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
groups(ba) <- list(NDE=NDE, DE=DE)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
res <- topCounts(ba, group="DE", normaliseData=T, number=nrow(data))
res <- res[rownames(data),]
q.value <- res$FDR.DE
ranking <- rank(q.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(res), res, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(res$ordering)
table(res$ordering)
table(res$ordering)/length(res$ordering)
obj <- (q.value < param_FDR)
sum(obj)
table(res$ordering[obj])
table(res$ordering[obj])/length(res$ordering[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
1.と同じですが、前処理のところが微妙に異なります。
具体的には、NDEとDEオブジェクト作成部分でfactor関数をつけていません。
また、文字列ベクトルから数値ベクトルになっています。こんな書き方でもOKという例です。約10分。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
param_samplesize <- 5000
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
NDE <- c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3))
DE <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
groups(ba) <- list(NDE=NDE, DE=DE)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
res <- topCounts(ba, group="DE", normaliseData=T, number=nrow(data))
res <- res[rownames(data),]
q.value <- res$FDR.DE
ranking <- rank(q.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(res), res, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(res$ordering)
table(res$ordering)
table(res$ordering)/length(res$ordering)
obj <- (q.value < param_FDR)
sum(obj)
table(res$ordering[obj])
table(res$ordering[obj])/length(res$ordering[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
Blekhman et al., Genome Res., 2010の20,689 genes×18 samplesのカウントデータです。
ヒトのメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3), チンパンジーのメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザルのメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
ここでは、ヒト(G1群)6サンプル, チンパンジー(G2群)6サンプル,
アカゲザル(G3群)6サンプルの3群間比較を行います。
既知のDEG情報はないのでAUC計算はできません。
約40分。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge3.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_samplesize <- 5000
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
NDE <- c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3))
DE <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
groups(ba) <- list(NDE=NDE, DE=DE)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
res <- topCounts(ba, group="DE", normaliseData=T, number=nrow(data))
res <- res[rownames(data),]
q.value <- res$FDR.DE
ranking <- rank(q.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(res), res, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(res$ordering)
table(res$ordering)
table(res$ordering)/length(res$ordering)
obj <- (q.value < param_FDR)
sum(obj)
table(res$ordering[obj])
table(res$ordering[obj])/length(res$ordering[obj])
3.と基本的に同じでparam_samplesizeで指定するリサンプリング回数を1/10にして実行時間を大幅に短縮しています。約4分。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge4.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_samplesize <- 500
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
NDE <- c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3))
DE <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
groups(ba) <- list(NDE=NDE, DE=DE)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
res <- topCounts(ba, group="DE", normaliseData=T, number=nrow(data))
res <- res[rownames(data),]
q.value <- res$FDR.DE
ranking <- rank(q.value)
tmp <- cbind(rownames(res), res, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
head(res$ordering)
table(res$ordering)
table(res$ordering)/length(res$ordering)
obj <- (q.value < param_FDR)
sum(obj)
table(res$ordering[obj])
table(res$ordering[obj])/length(res$ordering[obj])
例題1と基本的に同じですが、「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | EBSeq(Leng_2013)」
と同じ5種類の発現パターン(Pattern1, Pattern2, ..., Pattern5)を想定するやり方です。
出力形式もEBSeqに似せています。例題1は、Pattern1と5のみを想定するやり方です。約10分。AUC値は0.8783407ですね。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_samplesize <- 1000
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
Pat1 <- factor(c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3)))
Pat2 <- factor(c(rep(1, param_G1), rep(1, param_G2), rep(2, param_G3)))
Pat3 <- factor(c(rep(1, param_G1), rep(2, param_G2), rep(1, param_G3)))
Pat4 <- factor(c(rep(2, param_G1), rep(1, param_G2), rep(1, param_G3)))
Pat5 <- factor(c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3)))
groups(ba) <- list(Pattern1 = Pat1,
Pattern2 = Pat2,
Pattern3 = Pat3,
Pattern4 = Pat4,
Pattern5 = Pat5)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- paste("Pattern", max.col(out$PP), sep = "")
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
例題5と基本的に同じですが、例題1の実行結果中で見られるordering情報をranking列の左側に追加しています。
例えばgene_3はPattern4に属するとなっています。
Pattern4は「G1群が2、それ以外が1」を指定したPat4に対応するので「G1群で発現変動」です。
そのordering情報は"2>1"となっているので「G1群のほうが高発現のパターン」だというのがわかります。
同様に、gene_7はPattern3に属しています。
Pattern3は「G2群が2、それ以外が1」を指定したPat3に対応するので「G2群で発現変動」です。
そのordering情報は"1>2"となっているので「G2群のほうが低発現のパターン」だというのがわかります。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge6.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_samplesize <- 1000
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
Pat1 <- factor(c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3)))
Pat2 <- factor(c(rep(1, param_G1), rep(1, param_G2), rep(2, param_G3)))
Pat3 <- factor(c(rep(1, param_G1), rep(2, param_G2), rep(1, param_G3)))
Pat4 <- factor(c(rep(2, param_G1), rep(1, param_G2), rep(1, param_G3)))
Pat5 <- factor(c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3)))
groups(ba) <- list(Pattern1 = Pat1,
Pattern2 = Pat2,
Pattern3 = Pat3,
Pattern4 = Pat4,
Pattern5 = Pat5)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- paste("Pattern", max.col(out$PP), sep = "")
ranking <- rank(out$PP[, "Pattern1"])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
例題6と基本的に同じで、パターン名などを変更しているだけです。解釈のしやすさという点ではこちらのほうが優位かと思います。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge7.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_samplesize <- 1000
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_G1), rep("non", param_G2), rep("non", param_G3)))
DEG_G3 <- factor(c(rep("other", param_G1), rep("other", param_G2), rep("G3", param_G3)))
DEG_G2 <- factor(c(rep("other", param_G1), rep("G2", param_G2), rep("other", param_G3)))
DEG_G1 <- factor(c(rep("G1", param_G1), rep("other", param_G2), rep("other", param_G3)))
DEG_all <- factor(c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3)))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G3 = DEG_G3,
DEG_G2 = DEG_G2,
DEG_G1 = DEG_G1,
DEG_all = DEG_all)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- NULL
for(i in 1:nrow(out$PP)){
out$MAP <- append(out$MAP, colnames(out$PP)[max.col(out$PP)[i]])
}
ranking <- rank(out$PP[, "nonDEG"])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "nonDEG"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
8. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
サンプルデータ15のシミュレーションデータ作成から行うやり方です。
また、例題7と違って(このシミュレーションデータには存在しないので)「全ての群間で発現変動しているパターン」に相当するDEG_allを考慮しないやり方です。
AUC値は0.8747938ですね。
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8_confusion.txt"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
param_samplesize <- 1000
library(TCC)
library(baySeq)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_replicates[1]), rep("non", param_replicates[2]), rep("non", param_replicates[3])))
DEG_G1 <- factor(c(rep("G1", param_replicates[1]), rep("other", param_replicates[2]), rep("other", param_replicates[3])))
DEG_G2 <- factor(c(rep("other", param_replicates[1]), rep("G2", param_replicates[2]), rep("other", param_replicates[3])))
DEG_G3 <- factor(c(rep("other", param_replicates[1]), rep("other", param_replicates[2]), rep("G3", param_replicates[3])))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3)
libsizes(ba) <- getLibsizes(ba, estimationType="edgeR")
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- rank(out$PP[, param_narabi[1]])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
confusion <- matrix(0, nrow=length(param_narabi), ncol=length(param_narabi))
for(i in 1:length(param_narabi)){
confusion[i, 1] <- sum(out$MAP[nonDEG_posi] == param_narabi[i])
confusion[i, 2] <- sum(out$MAP[DEG_G1_posi] == param_narabi[i])
confusion[i, 3] <- sum(out$MAP[DEG_G2_posi] == param_narabi[i])
confusion[i, 4] <- sum(out$MAP[DEG_G3_posi] == param_narabi[i])
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 基礎 | edgeR(Robinson_2010)
TMM正規化(Robinson_2010)を実行したのち、
edgeRパッケージ中のGLM LRT法で発現変動遺伝子(Differentially Expressed Genes; DEGs)
検出を行うやり方(edgeR coupled with TMM normalization; TMM-edgeR)を示します。デフォルトのedgeRの手順に相当します。
ここでやっていることはANOVAのような「どこかの群間で発現に差がある遺伝子を検出」です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
2. サンプルデータ15の10,000 genes×9 samplesのシミュレーションデータ(data_hypodata_3vs3vs3.txt)と同じものをTCCパッケージ内部で作成する場合:
1.と同じ結果が得られます。
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
シミュレーションデータ(G1群2サンプル vs. G2群4サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge3.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3.と同じ結果が得られます。
out_f <- "hoge4.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
set.seed(1000)
tcc <- simulateReadCounts(Ngene = 10000, PDEG = 0.3,
DEG.assign = c(0.7, 0.2, 0.1),
DEG.foldchange = c(3, 10, 6),
replicates = c(param_G1, param_G2, param_G3))
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
calcAUCValue(tcc)
1.と基本的に同じで、出力のテキストファイルが正規化前のデータではなく正規化後のデータになっていて、発現変動順にソートしたものになっています。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
1.と基本的に同じですが、デザイン行列designと係数coefを明示的に作成および指定しています。
coefに入っている数値は、length(unique(data.cl))の値が3になるので、2:3、つまり2と3になります。
これがANOVA的などこかの群間で発現変動しているものを検出するときに指定する値です。
感覚的にはdesign[,2] - design[,3]をやっていることになり、「G1で-1, G2で0, G3で1」と全ての群で異なる数値が割り振られることになります。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge6.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
design <- model.matrix(~ as.factor(data.cl))
coef <- 2:length(unique(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=coef)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
3.と基本的に同じで、TCCパッケージを使わずにedgeRパッケージ中の関数のみで行うやり方です。TCCのほうが手順的にも簡便であることがわかります。
in_f <- "data_hypodata_2vs4vs3.txt"
out_f <- "hoge7.txt"
param_G1 <- 2
param_G2 <- 4
param_G3 <- 3
param_FDR <- 0.05
library(edgeR)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
d <- DGEList(data, group=data.cl)
d <- calcNormFactors(d)
design <- model.matrix(~ as.factor(data.cl))
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
coef <- 2:length(unique(data.cl))
lrt <- glmLRT(fit, coef=coef)
tmp <- topTags(lrt, n=nrow(data), sort.by="none")
p.value <- tmp$table$PValue
q.value <- tmp$table$FDR
result <- cbind(p.value, q.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(data), data, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
シミュレーションデータ(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_2000がG2群で5倍高発現、gene_2001〜gene_3000がG3群で5倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。このDEG or non-DEGの位置情報と実際のランキング結果情報を用いてAUC値の計算も行っています。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge8.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(TCC)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", iteration=0)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
8.と基本的に同じですが、TCCパッケージではなくedgeRパッケージ中の関数を利用しています。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge9.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_DEG <- 1:3000
library(edgeR)
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
d <- DGEList(data, group=data.cl)
d <- calcNormFactors(d)
design <- model.matrix(~ as.factor(data.cl))
d <- estimateGLMCommonDisp(d, design)
d <- estimateGLMTrendedDisp(d, design)
d <- estimateGLMTagwiseDisp(d, design)
fit <- glmFit(d, design)
coef <- 2:length(unique(data.cl))
lrt <- glmLRT(fit, coef=coef)
tmp <- topTags(lrt, n=nrow(data), sort.by="none")
p.value <- tmp$table$PValue
q.value <- tmp$table$FDR
result <- cbind(p.value, q.value)
sum(q.value < param_FDR)
ranking <- rank(p.value)
tmp <- cbind(rownames(data), data, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
obj <- rep(0, nrow(data))
obj[param_DEG] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)
「応用」でやりたいことは、例題1-7「指定した群間で発現に差がある遺伝子の検出」、そして例題8以降が「基礎」の結果と遺伝子間クラスタリングを組み合わせたパターン分類です。
例題1-7「指定した群間で発現に差がある遺伝子の検出」デザイン行列design中のparam_coefで指定した列を除くことでreduced modelを作成します。
TCCを用いたやり方を示します。
内部的にiDEGES/edgeR(Sun_2013)正規化を実行したのち、
edgeRパッケージ中のGLM LRT法で発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行っています。
TCC原著論文中のiDEGES/edgeR-edgeRという解析パイプラインに相当します。TCC原著論文(Sun et al., BMC Bioinformatics, 2013)では
3群間複製ありデータ用の推奨パイプラインを示していませんでしたが、多群間比較用の推奨ガイドライン提唱論文
(Tang et al., BMC Bioinformatics, 2015)
で推奨しているパイプライン"EEE-E"が"iDEGES/edgeR-edgeR"と同じものです。この2つの論文を引用し、安心してご利用ください。
例題8以降は、MBCluster.Seqも利用しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群4サンプル vs. G2群4サンプル vs. G3群4サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_2000がG2群で5倍高発現、gene_2001〜gene_3000がG3群で5倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。ANOVAと同じく、どこかの群間で発現変動しているものを検出するやり方(帰無仮説: G1 = G2 = G3)です。
出力ファイルのestimatedDEG列を眺めると、DEGに相当する最初の1行目から3000行目のところのほとんどが1,
それ以外の3001から10000行のほとんどが0になっていることからparam_coefでの指定方法が妥当であることが分かります。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge1.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_coef <- c(2, 3)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
design <- model.matrix(~ as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=param_coef)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
design
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「G1群 vs. G2群」の比較を行いたいときの指定方法(帰無仮説: G1 = G2)です。
出力ファイルのestimatedDEG列を眺めると、DEGに相当する最初の1行目から2000行目のところのほとんどが1である一方、
G3群で高発現DEGの2001から3000行目のほとんどが0となっていることから、param_coefでの指定方法が妥当であることが分かります。
「full modelに相当するデザイン行列designの2列目のパラメータを除いたものをreduced modelとする」という指定に相当します。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge2.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_coef <- 2
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
design <- model.matrix(~ as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=param_coef)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
design
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「G1群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G1 = G3)です。
出力ファイルのestimatedDEG列を眺めると、DEGに相当する最初の1-1000行目および2001-3000行目のところのほとんどが1である一方、
G2群で高発現DEGの1001から2000行目のほとんどが0となっていることから、param_coefでの指定方法が妥当であることが分かります。
「full modelに相当するデザイン行列designの3列目のパラメータを除いたものをreduced modelとする」という指定に相当します。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge3.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_coef <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
design <- model.matrix(~ as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=param_coef)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
design
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「G2群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G2 = G3)です。
出力ファイルのestimatedDEG列を眺めると、DEGに相当する最初の1001-3000行目のところのほとんどが1である一方、
G1群で高発現DEGの1から1000行目のほとんどが0となっていることから、param_contrastでの指定方法が妥当であることが分かります。
1-3.のようにparam_coefの枠組みではうまくG2 vs. G3をうまく表現できないので、コントラストという別の枠組みで指定しています。
思考回路としては、「a1*G1 + a2*G2 + a3*G3 = 0」として、この場合の目的である「G2 = G3という帰無仮説」を作成できるように、係数a1, a2, a3に適切な数値を代入することです。
ここではa1 = 0、a2 = -1, a3 = 1にすることで目的の帰無仮説を作成できます。
以下ではc(0, -1, 1)と指定していますが、
c(0, 1, -1)でも構いません。
理由はどちらでもG2 = G3を表現できているからです。
尚、full modelに相当するデザイン行列の作成手順も若干異なります。具体的には、model.matrix関数実行時に「0 + 」を追加しています。
これによって、最初の1列目が全て1になるようなG1群を基準にして作成したデザイン行列ではなく、各群が各列になるようにしています。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge4.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_contrast <- c(0, -1, 1)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
design <- model.matrix(~ 0 + as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, contrast=param_contrast)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
design
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「G1群 vs. G2群」の比較を行いたいときの指定方法(帰無仮説: G1 = G2)です。
出力ファイルのestimatedDEG列を眺めると、DEGに相当する最初の1行目から2000行目のところのほとんどが1である一方、
G3群で高発現DEGの2001から3000行目のほとんどが0となっていることから、param_contrastでの指定方法が妥当であることが分かります。
思考回路としては、「a1*G1 + a2*G2 + a3*G3 = 0」として、この場合の目的である「G1 = G2という帰無仮説」を作成できるように、係数a1, a2, a3に適切な数値を代入することです。
ここではa1 = 1、a2 = -1, a3 = 0にすることで目的の帰無仮説を作成できます。
以下ではc(1, -1, 0)と指定していますが、
c(-1, 1, 0)でも構いません。理由はどちらでもG1 = G2を表現できているからです。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge5.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_contrast <- c(1, -1, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
design <- model.matrix(~ 0 + as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, contrast=param_contrast)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
design
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「G1群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G1 = G3)です。
出力ファイルのestimatedDEG列を眺めると、DEGに相当する最初の1-1000行目および2001-3000行目のところのほとんどが1である一方、
G2群で高発現DEGの1001から2000行目のほとんどが0となっていることから、param_contrastでの指定方法が妥当であることが分かります。
思考回路としては、「a1*G1 + a2*G2 + a3*G3 = 0」として、この場合の目的である「G1 = G3という帰無仮説」を作成できるように、係数a1, a2, a3に適切な数値を代入することです。
ここではa1 = 1、a2 = 0, a3 = -1にすることで目的の帰無仮説を作成できます。
以下ではc(1, 0, -1)と指定していますが、
c(-1, 0, 1)でも構いません。理由はどちらでもG1 = G3を表現できているからです。
in_f <- "data_hypodata_4vs4vs4.txt"
out_f <- "hoge6.txt"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.1
param_contrast <- c(1, 0, -1)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
design <- model.matrix(~ 0 + as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, contrast=param_contrast)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
design
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題6までとは異なり、MBCluster.Seqを用いた遺伝子間クラスタリングを行った結果を合わせるやり方です。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge7.txt"
out_f2 <- "hoge7.png"
param_G1 <- 4
param_G2 <- 4
param_G3 <- 4
param_FDR <- 0.05
param_fig <- c(800, 500)
param_clust_num <- 10
library(TCC)
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
table(cls$cluster)
tmp <- cbind(rownames(tcc$count), normalized, result, cls$cluster)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
table(cls$cluster)
table(cls$cluster[q.value < 0.05])
table(cls$cluster[q.value < 0.10])
table(cls$cluster[q.value < 0.20])
table(cls$cluster[q.value < 0.30])
7.と基本的に同じで、入力ファイルが違うだけです。Blekhman et al., Genome Res., 2010の
20,689 genes×18 samplesのカウントデータです。ヒト(HS)、チンパンジー(PT)、アカゲザル(RM)の3生物種間比較です。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8.png"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_fig <- c(800, 500)
param_clust_num <- 10
library(TCC)
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
hoge <- RNASeq.Data(data, Normalizer=NULL,
Treatment=data.cl, GeneID=rownames(data))
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
table(cls$cluster)
tmp <- cbind(rownames(tcc$count), normalized, result, cls$cluster)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
table(cls$cluster)
table(cls$cluster[q.value < 0.05])
table(cls$cluster[q.value < 0.10])
table(cls$cluster[q.value < 0.20])
table(cls$cluster[q.value < 0.30])
8.と基本的に同じで、MBCluster.Seq実行時にTCC正規化後のデータを入力としています。
in_f <- "sample_blekhman_18.txt"
out_f1 <- "hoge9.txt"
out_f2 <- "hoge9.png"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_fig <- c(800, 500)
param_clust_num <- 10
library(TCC)
library(MBCluster.Seq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
hoge <- RNASeq.Data(data, Normalizer=size.factors,
Treatment=data.cl, GeneID=rownames(data))
set.seed(2016)
c0 <- KmeansPlus.RNASeq(data=hoge, nK=param_clust_num,
model="nbinom", print.steps=F)
cls <- Cluster.RNASeq(data=hoge, model="nbinom",
centers=c0$centers, method="EM")
tr <- Hybrid.Tree(data=hoge, model="nbinom",cluster=cls$cluster)
tmp <- cbind(rownames(tcc$count), normalized, result, cls$cluster)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plotHybrid.Tree(merge=tr, cluster=cls$cluster, logFC=hoge$logFC, tree.title=NULL)
dev.off()
table(cls$cluster)
table(cls$cluster[q.value < 0.05])
table(cls$cluster[q.value < 0.10])
table(cls$cluster[q.value < 0.20])
table(cls$cluster[q.value < 0.30])
- 多群間比較用の推奨ガイドライン提唱論文:Tang et al., BMC Bioinformatics, 2015
- TCC:Sun et al., BMC Bioinformatics, 2013
- TbT正規化法(TCCに実装されたDEGESアルゴリズム提唱論文):Kadota et al., Algorithms Mol. Biol., 2012
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR中のGLM論文:McCarthy et al., Nucleic Acids Res., 2012
- シリーズ Useful R 第7巻 トランスクリプトーム解析, 共立出版, 2014の4.2.2あたりにも解説あり
- MBCluster.Seq:Si et al., Bioinformatics, 2014
- k-means++:Arthur and Vassilvitskii, SODA '07 Proceedings of the eighteenth annual ACM-SIAM symposium on Discrete algorithms, 2007
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | Blekhmanデータ | TCC(Sun_2013)
「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC(Sun_2013)」
と基本的に同じです。Blekhman et al., Genome Res., 2010の公共カウントデータ解析に特化させて、
TCCを用いた様々な例題を示します。
入力は全てサンプルデータ42の20,689 genes×18 samplesのリアルカウントデータ
(sample_blekhman_18.txt)です。
ヒトHomo sapiens; HS)のメス3サンプル(HSF1-3)とオス3サンプル(HSM1-3),
チンパンジー(Pan troglodytes; PT)のメス3サンプル(PTF1-3)とオス3サンプル(PTM1-3),
アカゲザル(Rhesus macaque; RM)のメス3サンプル(RMF1-3)とオス3サンプル(RMM1-3)の並びになっています。
つまり、以下のような感じです。FはFemale(メス)、MはMale(オス)を表します。
ヒト(1-6列目): HSF1, HSF2, HSF3, HSM1, HSM2, and HSM3
チンパンジー(7-12列目): PTF1, PTF2, PTF3, PTM1, PTM2, and PTM3
アカゲザル(13-18列目): RMF1, RMF2, RMF3, RMM1, RMM2, and RMM3
2022年9月2日にいただいた情報として、一部の例題で挙動がおかしいようです。少なくとも例題4でエラーが出ることを私も確認済みです。この原因はTCCパッケージが内部的に用いているedgeRの仕様変更に起因します。半年ほど前まではうまく動いていたようですが、多群間比較のpost-hoc testは組み合わせも多数あるので作業が煩雑です。それゆえ、多群間比較の場合は「解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+baySeq(Osabe_2019)」の例題8以降を参考にして解析するようにしてください。こちらのやり方だと、どの群間で発現変動しているかといった発現パターン分類までone-stopでやってくれます(この推奨パイプラインの原著論文はOsabe et al., Bioinform. Biol. Insight, 2019です)。(2022/09/03)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. ANOVA的な解析の場合:
どこかの群間で発現変動しているものを検出するやり方(帰無仮説: G1 = G2 = G3)です。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge1.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_coef <- c(2, 3)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
design <- model.matrix(~ as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=param_coef)
result <- getResult(tcc, sort=FALSE)
design
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
2. post-hoc testで「G1群 vs. G2群」の比較の場合:
「G1群 vs. G2群」の比較を行いたいときの指定方法(帰無仮説: G1 = G2)です。
「full modelに相当するデザイン行列designの2列目のパラメータを除いたものをreduced modelとする」という指定に相当します。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge2.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_coef <- 2
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
design <- model.matrix(~ as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=param_coef)
result <- getResult(tcc, sort=FALSE)
design
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
3. post-hoc testで「G1群 vs. G3群」の比較の場合:
「G1群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G1 = G3)です。
「full modelに相当するデザイン行列designの3列目のパラメータを除いたものをreduced modelとする」という指定に相当します。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge3.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_coef <- 3
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
design <- model.matrix(~ as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, coef=param_coef)
result <- getResult(tcc, sort=FALSE)
design
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
4. post-hoc testで「G2群 vs. G3群」の比較の場合:
「G2群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G2 = G3)です。
param_coefの枠組みではうまくG2 vs. G3をうまく表現できないので、コントラストという別の枠組みで指定しています。
思考回路としては、「a1*G1 + a2*G2 + a3*G3 = 0」として、この場合の目的である「G2 = G3という帰無仮説」を作成できるように、係数a1, a2, a3に適切な数値を代入することです。
ここではa1 = 0、a2 = -1, a3 = 1にすることで目的の帰無仮説を作成できます。
以下ではc(0, -1, 1)と指定していますが、
c(0, 1, -1)でも構いません。
理由はどちらでもG2 = G3を表現できているからです。
尚、full modelに相当するデザイン行列の作成手順も若干異なります。具体的には、model.matrix関数実行時に「0 + 」を追加しています。
これによって、最初の1列目が全て1になるようなG1群を基準にして作成したデザイン行列ではなく、各群が各列になるようにしています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge4.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_contrast <- c(0, -1, 1)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
#design <- model.matrix(~ as.factor(data.cl))
design <- model.matrix(~ 0 + as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, contrast=param_contrast)
result <- getResult(tcc, sort=FALSE)
design
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
5. post-hoc testで「G1群 vs. G2群」の比較の場合:
コントラストで「G1群 vs. G2群」の比較を行いたいときの指定方法(帰無仮説: G1 = G2)です。
ここではa1 = 1、a2 = -1, a3 = 0にすることで目的の帰無仮説を作成できます。
以下ではc(1, -1, 0)と指定していますが、
c(-1, 1, 0)でも構いません。
理由はどちらでもG1 = G2を表現できているからです。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge5.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_contrast <- c(1, -1, 0)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
#design <- model.matrix(~ as.factor(data.cl))
design <- model.matrix(~ 0 + as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, contrast=param_contrast)
result <- getResult(tcc, sort=FALSE)
design
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
6. post-hoc testで「G1群 vs. G3群」の比較の場合:
コントラストで「G1群 vs. G3群」の比較を行いたいときの指定方法(帰無仮説: G1 = G3)です。
ここではa1 = 1、a2 = 0, a3 = -1にすることで目的の帰無仮説を作成できます。
以下ではc(1, 0, -1)と指定していますが、
c(-1, 0, 1)でも構いません。
理由はどちらでもG1 = G3を表現できているからです。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge6.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.05
param_contrast <- c(1, 0, -1)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
#design <- model.matrix(~ as.factor(data.cl))
design <- model.matrix(~ 0 + as.factor(data.cl))
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR,
design=design, contrast=param_contrast)
result <- getResult(tcc, sort=FALSE)
design
tmp <- cbind(rownames(tcc$count), normalized, result)
tmp <- tmp[order(tmp$rank),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.20)
sum(q.value < 0.30)
- 多群間比較用の推奨ガイドライン提唱論文:Tang et al., BMC Bioinformatics, 2015
- TCC:Sun et al., BMC Bioinformatics, 2013
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR中のGLM論文:McCarthy et al., Nucleic Acids Res., 2012
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+baySeq(Osabe_2019)
TCCとbaySeq
を組み合わせたやり方を示します。例題8以降が推奨パイプライン(Osabe et al., Bioinform. Biol. Insight, 2019)です(2019年7月10日追記)。
例題1-7までは、「TCC中のiDEGES/edgeR正規化で得られた正規化係数をbaySeqに与えた解析パイプライン」です。
TCC原著論文中のiDEGES/edgeR-baySeqという解析パイプラインに相当し、
多群間比較用の推奨ガイドライン提唱論文(Tang et al., 2015)の表記法に従うとEEE-bに相当します。
その後、このパイプライン(EEE-b)は、TCCのデフォルトの解析パイプライン(EEE-E)よりも全体的な発現変動のランキングの点で劣っていることが判明しました(Osabe et al., 2019)。
しかし、EEE-Eはどこかの群間で発現変動しているというANOVA的な結果までしか返さないのに対して、EEE-bは発現変動パターンの割当て(や分類)の点で優位です。
理由は、TCCやedgeRやDESeq2を用いて3群間比較で発現変動パターンの割当まで行う場合、「G1 vs. G2、G1 vs. G3、G2 vs. G3の3通りの2群間比較を独立に行ってから、
その結果に基づいて発現変動パターンを構築する」とか、あるいは
「(G1+G2) vs. G3、(G1+G3) vs. G2、(G2+G3) vs. G1を行ってから構築する」などやろうと思えばやれないことはないが現実には結構大変だからです。
例題8は、全体的な発現変動の度合い(ANOVA的などこかの群間で発現変動している度合いでのランキング)をEEE-Eで行い、
発現変動パターンの同定をEEE-b(長部らの原著論文中ではEEE-baySEqと表記)で行った結果を出力しています。
以下は2020年07月29日に追加した情報。入力ファイルに実数が含まれるとうまく動きませんのでご注意ください。
例えば、STAR-RSEMで作成したものでexpected_countをそのまま入力として与えたい場合は、
入力ファイル読み込み後のオブジェクトに対してround関数を実行するなどすればよいです(茂木朋貴 氏提供情報)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(2015)の前に#を入れましょう。
つまり「set.seed(2015)」->「#set.seed(2015)」です。
2015はタネ番号なので3や496などでも構いません。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
param_samplesize <- 1000
library(TCC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
NDE <- c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3))
DE <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
groups(ba) <- list(NDE=NDE, DE=DE)
ba@annotation <- data.frame(name = tcc$gene_id)
libsizes(ba) <- colSums(data)*tcc$norm.factors
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
res <- topCounts(ba, group="DE", normaliseData=T, number=nrow(data))
rownames(res) <- res$name
res <- res[rownames(data),]
q.value <- res$FDR.DE
ranking <- rank(q.value)
sum(q.value < param_FDR)
tmp <- cbind(rownames(res), res, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(res$DE)
table(res$DE)
table(res$DE)/length(res$DE)
obj <- (q.value < param_FDR)
sum(obj)
table(res$DE[obj])
table(res$DE[obj])/length(res$DE[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(2015)の前に#を入れましょう。
つまり「set.seed(2015)」->「#set.seed(2015)」です。
2015はタネ番号なので3や496などでも構いません。
出力ファイルのout$MAP列にはPattern1からPattern4までのいずれかが表示されていますが、
「Pattern1はnon-DEG、Pattern2はG1群で発現変動、Pattern3はG2群で発現変動、Pattern4はG3群で発現変動」
だと読み解きます。orderings列には、発現変動の方向についての情報が記載されています。Pattern1はnon-DEGなので情報はありません。
Pattern2から4までについて「1<2または2>1」の情報が記載されています。例えばPattern2はG1群に着目していますので、
着目している群のほうが高発現であれば2>1が、低発現であれば1<2が表示されます。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
param_samplesize <- 1000
library(TCC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
Pat1 <- factor(c(rep(1, param_G1), rep(1, param_G2), rep(1, param_G3)))
Pat2 <- factor(c(rep(2, param_G1), rep(1, param_G2), rep(1, param_G3)))
Pat3 <- factor(c(rep(1, param_G1), rep(2, param_G2), rep(1, param_G3)))
Pat4 <- factor(c(rep(1, param_G1), rep(1, param_G2), rep(2, param_G3)))
groups(ba) <- list(Pattern1 = Pat1,
Pattern2 = Pat2,
Pattern3 = Pat3,
Pattern4 = Pat4)
ba@annotation <- data.frame(name = tcc$gene_id)
libsizes(ba) <- colSums(data)*tcc$norm.factors
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- paste("Pattern", max.col(out$PP), sep = "")
ranking <- rank(out$PP[, "Pattern1"])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
3. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
サンプルデータ15のシミュレーションデータ作成から行うやり方です。
out_f <- "hoge3.txt"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
param_samplesize <- 1000
library(TCC)
library(baySeq)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
Pat1 <- factor(c(rep(1, param_replicates[1]), rep(1, param_replicates[2]), rep(1, param_replicates[3])))
Pat2 <- factor(c(rep(2, param_replicates[1]), rep(1, param_replicates[2]), rep(1, param_replicates[3])))
Pat3 <- factor(c(rep(1, param_replicates[1]), rep(2, param_replicates[2]), rep(1, param_replicates[3])))
Pat4 <- factor(c(rep(1, param_replicates[1]), rep(1, param_replicates[2]), rep(2, param_replicates[3])))
groups(ba) <- list(Pattern1 = Pat1,
Pattern2 = Pat2,
Pattern3 = Pat3,
Pattern4 = Pat4)
libsizes(ba) <- colSums(data)*tcc$norm.factors
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- paste("Pattern", max.col(out$PP), sep = "")
ranking <- rank(out$PP[, "Pattern1"])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
4. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題3と基本的に同じで、パターン名などを変更しているだけです。解釈のしやすさという点ではこちらのほうが優位かと思います。
out_f <- "hoge4.txt"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
param_samplesize <- 1000
library(TCC)
library(baySeq)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_replicates[1]), rep("non", param_replicates[2]), rep("non", param_replicates[3])))
DEG_G1 <- factor(c(rep("G1", param_replicates[1]), rep("other", param_replicates[2]), rep("other", param_replicates[3])))
DEG_G2 <- factor(c(rep("other", param_replicates[1]), rep("G2", param_replicates[2]), rep("other", param_replicates[3])))
DEG_G3 <- factor(c(rep("other", param_replicates[1]), rep("other", param_replicates[2]), rep("G3", param_replicates[3])))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3)
libsizes(ba) <- colSums(data)*tcc$norm.factors
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- rank(out$PP[, param_narabi[1]])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, param_narabi[1]] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
5. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題4と基本的に同じです。混同行列(confusion matrix)情報のファイルも出力しています。
AUC値は0.8917377ですね。
out_f1 <- "hoge5.txt"
out_f2 <- "hoge5_confusion.txt"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
param_samplesize <- 1000
library(TCC)
library(baySeq)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_replicates[1]), rep("non", param_replicates[2]), rep("non", param_replicates[3])))
DEG_G1 <- factor(c(rep("G1", param_replicates[1]), rep("other", param_replicates[2]), rep("other", param_replicates[3])))
DEG_G2 <- factor(c(rep("other", param_replicates[1]), rep("G2", param_replicates[2]), rep("other", param_replicates[3])))
DEG_G3 <- factor(c(rep("other", param_replicates[1]), rep("other", param_replicates[2]), rep("G3", param_replicates[3])))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3)
libsizes(ba) <- colSums(data)*tcc$norm.factors
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- rank(out$PP[, param_narabi[1]])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
hoge <- param_Ngene*param_PDEG
DEG_G1_posi <- 1:(hoge*param_DEGassign[1])
DEG_G2_posi <- (hoge*param_DEGassign[1]+1):(hoge*sum(param_DEGassign[1:2]))
DEG_G3_posi <- (hoge*sum(param_DEGassign[1:2])+1):hoge
nonDEG_posi <- (hoge+1):param_Ngene
confusion <- matrix(0, nrow=length(param_narabi), ncol=length(param_narabi))
for(i in 1:length(param_narabi)){
confusion[i, 1] <- sum(out$MAP[nonDEG_posi] == param_narabi[i])
confusion[i, 2] <- sum(out$MAP[DEG_G1_posi] == param_narabi[i])
confusion[i, 3] <- sum(out$MAP[DEG_G2_posi] == param_narabi[i])
confusion[i, 4] <- sum(out$MAP[DEG_G3_posi] == param_narabi[i])
}
confusion <- cbind(confusion, rowSums(confusion))
confusion <- rbind(confusion, colSums(confusion))
colnames(confusion) <- c(param_narabi, "Total")
rownames(confusion) <- c(param_narabi, "Total")
confusion
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
tmp <- cbind(rownames(confusion), confusion)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
library(ROC)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(2015)の前に#を入れましょう。
つまり「set.seed(2015)」->「#set.seed(2015)」です。
2015はタネ番号なので3や496などでも構いません。
発現パターンとして、nonDEG(発現変動なし), DEG_G1(G1群で発現変動), DEG_G2(G2群で発現変動), DEG_G3(G3群で発現変動)の計4パターンのみを考慮するやり方です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge6.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3")
param_samplesize <- 1000
library(TCC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_G1), rep("non", param_G2), rep("non", param_G3)))
DEG_G1 <- factor(c(rep("G1", param_G1), rep("other", param_G2), rep("other", param_G3)))
DEG_G2 <- factor(c(rep("other", param_G1), rep("G2", param_G2), rep("other", param_G3)))
DEG_G3 <- factor(c(rep("other", param_G1), rep("other", param_G2), rep("G3", param_G3)))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3)
libsizes(ba) <- colSums(data)*tcc$norm.factors
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- rank(out$PP[, param_narabi[1]])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, param_narabi[1]] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(2015)の前に#を入れましょう。
つまり「set.seed(2015)」->「#set.seed(2015)」です。
2015はタネ番号なので3や496などでも構いません。
発現パターンとして、nonDEG(発現変動なし), DEG_G1(G1群で発現変動), DEG_G2(G2群で発現変動), DEG_G3(G3群で発現変動), DEGall(全ての群で発現変動)の計5パターンを考慮するやり方です。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge7.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3", "DEGall")
param_samplesize <- 1000
library(TCC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_G1), rep("non", param_G2), rep("non", param_G3)))
DEG_G1 <- factor(c(rep("G1", param_G1), rep("other", param_G2), rep("other", param_G3)))
DEG_G2 <- factor(c(rep("other", param_G1), rep("G2", param_G2), rep("other", param_G3)))
DEG_G3 <- factor(c(rep("other", param_G1), rep("other", param_G2), rep("G3", param_G3)))
DEGall <- factor(c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3)))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3,
DEGall = DEGall)
libsizes(ba) <- colSums(data)*tcc$norm.factors
set.seed(2015)
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- rank(out$PP[, param_narabi[1]])
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, param_narabi[1]] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
例題7と似ていますが、全体的な発現変動の度合い(ANOVA的などこかの群間で発現変動している度合いでのランキング)をEEE-Eで行い、
発現変動パターンの同定をEEE-b(長部らの原著論文中ではEEE-baySEqと表記)で行った結果を出力しています
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge8.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3", "DEGall")
param_samplesize <- 1000
library(TCC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_G1), rep("non", param_G2), rep("non", param_G3)))
DEG_G1 <- factor(c(rep("G1", param_G1), rep("other", param_G2), rep("other", param_G3)))
DEG_G2 <- factor(c(rep("other", param_G1), rep("G2", param_G2), rep("other", param_G3)))
DEG_G3 <- factor(c(rep("other", param_G1), rep("other", param_G2), rep("G3", param_G3)))
DEGall <- factor(c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3)))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3,
DEGall = DEGall)
set.seed(2015)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
libsizes(ba) <- colSums(data)*tcc$norm.factors
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- tcc$stat$rank
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking, result$q.value)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (tcc$stat$q.value < param_FDR)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
例題8とは基本的に入力ファイルが異なるだけです。入力ファイルは、1~1,000行目がG1群で4倍高発現パターン(DEG_G1)、
1,001~2,000行目がG2群で4倍高発現パターン(DEG_G2)、2,001~3,000行目がG3群で4倍高発現パターン(DEG_G3)、残りがnon-DEGです。
他は、TCC-GUI (Su et al., 2019)の
デフォルトのFDR閾値(= 0.10)に合わせています。
in_f <- "Simulation_3group_sheep.txt"
out_f <- "hoge9.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.10
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3", "DEGall")
param_samplesize <- 1000
library(TCC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_G1), rep("non", param_G2), rep("non", param_G3)))
DEG_G1 <- factor(c(rep("G1", param_G1), rep("other", param_G2), rep("other", param_G3)))
DEG_G2 <- factor(c(rep("other", param_G1), rep("G2", param_G2), rep("other", param_G3)))
DEG_G3 <- factor(c(rep("other", param_G1), rep("other", param_G2), rep("G3", param_G3)))
DEGall <- factor(c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3)))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3,
DEGall = DEGall)
set.seed(2015)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
libsizes(ba) <- colSums(data)*tcc$norm.factors
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- tcc$stat$rank
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking, result$q.value)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (tcc$stat$q.value < param_FDR)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
例題9とは基本的に入力ファイルが異なるだけです。
サンプルデータ42で作成したリアルデータです。
HS(ヒト)6 samples、PT(チンパンジー)6 samples、RM(アカゲザル)6 samplesの3生物種間比較用です。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge10.txt"
param_G1 <- 6
param_G2 <- 6
param_G3 <- 6
param_FDR <- 0.10
param_narabi <- c("nonDEG", "DEG_G1", "DEG_G2", "DEG_G3", "DEGall")
param_samplesize <- 1000
library(TCC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
ba <- new("countData", data=as.matrix(data), replicates=data.cl)
nonDEG <- factor(c(rep("non", param_G1), rep("non", param_G2), rep("non", param_G3)))
DEG_G1 <- factor(c(rep("G1", param_G1), rep("other", param_G2), rep("other", param_G3)))
DEG_G2 <- factor(c(rep("other", param_G1), rep("G2", param_G2), rep("other", param_G3)))
DEG_G3 <- factor(c(rep("other", param_G1), rep("other", param_G2), rep("G3", param_G3)))
DEGall <- factor(c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3)))
groups(ba) <- list(nonDEG = nonDEG,
DEG_G1 = DEG_G1,
DEG_G2 = DEG_G2,
DEG_G3 = DEG_G3,
DEGall = DEGall)
set.seed(2015)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
libsizes(ba) <- colSums(data)*tcc$norm.factors
ba <- getPriors.NB(ba, samplesize=param_samplesize, estimation="QL", cl=NULL)
ba <- getLikelihoods(ba, pET="BIC", nullData=FALSE, cl=NULL)
out <- list()
out$PP <- exp(ba@posteriors)
out$MAP <- param_narabi[max.col(out$PP)]
ranking <- tcc$stat$rank
orderings <- NULL
for(i in 1:length(out$MAP)){
orderings <- append(orderings, as.character(ba@orderings[i, max.col(out$PP)[i]]))
}
tmp <- cbind(rownames(data), data, out$PP, out$MAP, orderings, ranking, result$q.value)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (tcc$stat$q.value < param_FDR)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
- Osabe et al., Bioinform. Biol. Insight, 2019
- TCC-GUI:Su et al., BMC Res. Notes, 2019
- 多群間比較用の推奨ガイドライン提唱論文:Tang et al., BMC Bioinformatics, 2015
- TCC:Sun et al., BMC Bioinformatics, 2013
- TbT正規化法(TCCに実装されたDEGESアルゴリズム提唱論文):Kadota et al., Algorithms Mol. Biol., 2012
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR中のGLM論文:McCarthy et al., Nucleic Acids Res., 2012
- baySeq:Hardcastle and Kelly, BMC Bioinformatics, 2010
解析 | 発現変動 | 3群間 | 対応なし | 複製あり | 応用 | TCC+EBSeq(Osabe_2019)
TCCとEBSeq
を組み合わせたやり方を示します。
例題5以降がEBSeqを使う場合の推奨パイプライン(Osabe et al., Bioinform. Biol. Insight, 2019)です(2019年7月10日追記)。
例題1-4までは、「TCC中のiDEGES/edgeR正規化で得られた正規化係数をEBSeqに与えた解析パイプライン」です。
TCC原著論文の表記法に従うと、iDEGES/edgeR-EBSeqという解析パイプラインに相当します。
Tang et al., 2015の表記法では、、、EEE-EBSeqですね。
出力結果ファイルを眺めると
Pattern1, Pattern2, ..., Pattern5までの列のところに遺伝子ごとのPosterior Probability (PP)値が与えられています。
PP値が一番大きい(つまり1に近い)パターンに、その遺伝子が属するということになります:
Pattern1は(1, 1, 1)なのでnon-DEGの発現パターン
Pattern2は(1, 1, 2)なのでG3群で発現変動(高発現または低発現)
Pattern3は(1, 2, 1)なのでG2群で発現変動
Pattern4は(1, 2, 2)なのでG1群で発現変動
Pattern5は(1, 2, 3)なので、全ての群間で発現変動
一番右側のout$MAP列を見る方が手っ取り早いかもしれません。どの遺伝子がどのパターンに属するかが一目瞭然です。
ranking列は、Pattern1のPP値に基づいています。Pattern1のPP値の低さはnon-DEGである確率の低さを意味するからです。
尚、試行(trial)ごとに得られる数値が変わるようにしたい場合はset.seed(2015)の前に#を入れましょう。
つまり「set.seed(2015)」->「#set.seed(2015)」です。
2015はタネ番号なので3や496などでも構いません。
2018年8月12日に、本来「size.factors <- ef.libsizes/mean(ef.libsizes)」と書くべきところを、どこでどう間違えたのか
「size.factors <- mean(ef.libsizes)/ef.libsizes」となっていたことが判明したので修正しました(長部 高之 氏提供情報)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)、
そしてgene_3001〜gene_10000までがnon-DEGであることが既知です。
従って、Pattern1が70%、Pattern2が3%、Pattern3が6%、Pattern4が21%、Pattern5が0%が正解になります。
AUC値は0.8534662ですね。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
library(TCC)
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
PosParti <- GetPatterns(data.cl)
Parti <- PosParti
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
例題1との違いは、(このシミュレーションデータには存在しないので)「全ての群間で発現変動しているパターン」に相当するPattern5を考慮しないやり方です。
AUC値は0.8534662ですね。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
library(TCC)
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2), rep("G3", param_G3))
PosParti <- GetPatterns(data.cl)
obj <- (rownames(PosParti) != "Pattern5")
Parti <- PosParti[obj, ]
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- rep(0, nrow(data))
obj[1:3000] <- 1
AUC(rocdemo.sca(truth=obj, data=-ranking))
3. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
サンプルデータ15のシミュレーションデータ作成から行うやり方です。
AUC値は0.8534662です。
out_f <- "hoge3.txt"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
library(TCC)
library(EBSeq)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
data.cl <- c(rep("G1", param_replicates[1]), rep("G2", param_replicates[2]), rep("G3", param_replicates[3]))
PosParti <- GetPatterns(data.cl)
obj <- (rownames(PosParti) != "Pattern5")
Parti <- PosParti[obj, ]
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
4. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題3と基本的に同じですが、混同行列(confusion matrix)情報のファイルも出力しています。
out_f1 <- "hoge4.txt"
out_f2 <- "hoge4_confusion.txt"
param_replicates <- c(3, 3, 3)
param_Ngene <- 10000
param_PDEG <- 0.3
param_FC <- c(3, 10, 6)
param_DEGassign <- c(0.7, 0.2, 0.1)
library(TCC)
library(EBSeq)
set.seed(1000)
tcc <- simulateReadCounts(Ngene=param_Ngene,
PDEG=param_PDEG,
DEG.assign=param_DEGassign,
DEG.foldchange=param_FC,
replicates=param_replicates)
data <- tcc$count
data.cl <- tcc$group$group
dim(data)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
data.cl <- c(rep("G1", param_replicates[1]), rep("G2", param_replicates[2]), rep("G3", param_replicates[3]))
PosParti <- GetPatterns(data.cl)
obj <- (rownames(PosParti) != "Pattern5")
Parti <- PosParti[obj, ]
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
ranking <- rank(out$PP[, "Pattern1"])
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (out$PP[, "Pattern1"] < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
library(ROC)
obj <- as.numeric(tcc$simulation$trueDEG != 0)
AUC(rocdemo.sca(truth=obj, data=-ranking))
5. サンプルデータ15の10,000 genes×9 samplesのカウントデータ生成から行う場合:
例題1-4までとは異なり、全体的な発現変動の度合いでのランキングはEEE-Eのパイプラインを利用しています。
発現変動パターンの同定部分がEEE-EBSeqです。
in_f <- "data_hypodata_3vs3vs3.txt"
out_f <- "hoge5.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_FDR <- 0.05
library(TCC)
library(EBSeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
norm.factors <- tcc$norm.factors
ef.libsizes <- colSums(data)*norm.factors
size.factors <- ef.libsizes/mean(ef.libsizes)
size.factors
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
ranking <- tcc$stat$rank
sum(tcc$stat$q.value < param_FDR)
Parti <- GetPatterns(data.cl)
Parti
hoge <- EBMultiTest(as.matrix(data), NgVector=NULL,
Conditions=data.cl, AllParti=Parti,
sizeFactors=size.factors, maxround=5,
Qtrm=1.0, QtrmCut=-1)
out <- GetMultiPP(hoge)
tmp <- cbind(rownames(data), data, out$PP, out$MAP, ranking)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
out$Patterns
head(out$MAP)
table(out$MAP)
table(out$MAP)/length(out$MAP)
obj <- (tcc$stat$q.value < 0.05)
sum(obj)
table(out$MAP[obj])
table(out$MAP[obj])/length(out$MAP[obj])
- Osabe et al., Bioinform. Biol. Insight, 2019
- 多群間比較用の推奨ガイドライン提唱論文:Tang et al., BMC Bioinformatics, 2015
- TCC:Sun et al., BMC Bioinformatics, 2013
- TbT正規化法(TCCに実装されたDEGESアルゴリズム提唱論文):Kadota et al., Algorithms Mol. Biol., 2012
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR中のGLM論文:McCarthy et al., Nucleic Acids Res., 2012
- EBSeq:Leng et al., Bioinformatics, 2013
解析 | 発現変動 | 3群間 | 対応なし | 複製なし | DESeq2(Love_2014)
DESeq2パッケージ
(Love et al., Genome Biol., 2014)を用いるやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル vs. G3群1サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
約1分かかります。
in_f <- "data_hypodata_1vs1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
param_G3 <- 1
library(DESeq2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
d <- DESeq(d)
#d <- estimateSizeFactors(d)
##sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))
#d <- estimateDispersions(d)
#d <- nbinomLRT(d, full= ~condition, reduced= ~1)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
tmp <- cbind(rownames(data), data, p.value, q.value, ranking)
tmp <- tmp[order(ranking),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
sum(q.value < 0.80)
sum(p.adjust(p.value, method="BH") < 0.05)
sum(p.adjust(p.value, method="BH") < 0.10)
sum(p.adjust(p.value, method="BH") < 0.30)
sum(p.adjust(p.value, method="BH") < 0.50)
sum(p.adjust(p.value, method="BH") < 0.70)
sum(p.adjust(p.value, method="BH") < 0.80)
20,689 genes×18 samplesのカウントデータで、ヒト(HS)、チンパンジー(PT)、アカゲザル(RM)の3生物種間比較です
(Blekhman et al., Genome Res., 2010)。
1, 7, 13列目のデータのみ抽出して、
反復なし3群間比較(HSF1 vs. PTF1 vs. RMF1)としています。
正規化後のデータで発現変動順にソートした結果を出力しています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge2.txt"
param_subset <- c(1, 7, 13)
param_G1 <- 1
param_G2 <- 1
param_G3 <- 1
library(DESeq2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
colData <- data.frame(condition=as.factor(data.cl))
d <- DESeqDataSetFromMatrix(countData=data, colData=colData, design=~condition)
d <- DESeq(d)
#d <- estimateSizeFactors(d)
##sizeFactors(d) <- sizeFactors(d)/mean(sizeFactors(d))
#d <- estimateDispersions(d)
#d <- nbinomLRT(d, full= ~condition, reduced= ~1)
normalized <- counts(d, normalized=T)
tmp <- results(d)
p.value <- tmp$pvalue
p.value[is.na(p.value)] <- 1
q.value <- tmp$padj
q.value[is.na(q.value)] <- 1
ranking <- rank(p.value)
tmp <- cbind(rownames(data), normalized, p.value, q.value, ranking)
tmp <- tmp[order(ranking),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
sum(q.value < 0.80)
sum(p.adjust(p.value, method="BH") < 0.05)
sum(p.adjust(p.value, method="BH") < 0.10)
sum(p.adjust(p.value, method="BH") < 0.30)
sum(p.adjust(p.value, method="BH") < 0.50)
sum(p.adjust(p.value, method="BH") < 0.70)
sum(p.adjust(p.value, method="BH") < 0.80)
解析 | 発現変動 | 3群間 | 対応なし | 複製なし | TCC(Sun_2013)
TCCを用いたやり方を示します。
内部的にDESeqパッケージ中の関数を利用して、
複製なしデータに対応済みのDESeq2の通常の手順を複数回繰り返す(DEGES-based normalization; Kadota et al., 2012)
ことでより正確なデータ正規化が実現された発現変動解析結果を得ることができます。
2019年7月11日にこの項目内で明示的にDESeq2と指定していた部分をDESeqに変更しました。理由はDESeq2を使うとエラーが出るようになったからです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群1サンプル vs. G2群1サンプル vs. G3群1サンプル)です。
gene_1〜gene_3000までがDEG (gene_1〜gene_2100がG1群で3倍高発現、gene_2101〜gene_2700がG2群で10倍高発現、gene_2701〜gene_3000がG3群で6倍高発現)
gene_3001〜gene_10000までがnon-DEGであることが既知です。
約1分かかります。
2018年の秋ごろのリリースでDESeq2が反復なし非対応になったので、TCC側がまだ対応できておらずエラーとなります(2019年4月26日追加)。
in_f <- "data_hypodata_1vs1vs1.txt"
out_f <- "hoge1.txt"
param_G1 <- 1
param_G2 <- 1
param_G3 <- 1
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, iteration=3)
tcc <- estimateDE(tcc, FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
明示的にDESeqを指定しています。約1分。
in_f <- "data_hypodata_1vs1vs1.txt"
out_f <- "hoge2.txt"
param_G1 <- 1
param_G2 <- 1
param_G3 <- 1
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq",
test.method="deseq", iteration=3)
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
20,689 genes×18 samplesのカウントデータで、ヒト(HS)、チンパンジー(PT)、アカゲザル(RM)の3生物種間比較です
(Blekhman et al., Genome Res., 2010)。
1, 7, 13列目のデータのみ抽出して、
反復なし3群間比較(HSF1 vs. PTF1 vs. RMF1)としています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge3.txt"
param_subset <- c(1, 7, 13)
param_G1 <- 1
param_G2 <- 1
param_G3 <- 1
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
colnames(data)
tcc <- calcNormFactors(tcc, norm.method="deseq",
test.method="deseq", iteration=3)
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
q.value <- tcc$stat$q.value
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
20,689 genes×18 samplesのカウントデータで、ヒト(HS)、チンパンジー(PT)、アカゲザル(RM)の3生物種間比較です
(Blekhman et al., Genome Res., 2010)。
1, 7, 13列目のデータのみ抽出して、
反復なし3群間比較(HSF1 vs. PTF1 vs. RMF1)としています。
例題3と基本的に同じですが、正規化後のデータで発現変動順にソートした結果を出力しています。
in_f <- "sample_blekhman_18.txt"
out_f <- "hoge4.txt"
param_subset <- c(1, 7, 13)
param_G1 <- 1
param_G2 <- 1
param_G3 <- 1
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data <- data[,param_subset]
data.cl <- c(rep(1, param_G1), rep(2, param_G2), rep(3, param_G3))
tcc <- new("TCC", data, data.cl)
colnames(data)
tcc <- calcNormFactors(tcc, norm.method="deseq",
test.method="deseq", iteration=3)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="deseq", FDR=param_FDR)
p.value <- tcc$stat$p.value
p.value[is.na(p.value)] <- 1
ranking <- rank(p.value)
q.value <- tcc$stat$q.value
tmp <- cbind(rownames(tcc$count), normalized, p.value, q.value, ranking)
tmp <- tmp[order(ranking),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
sum(q.value < 0.05)
sum(q.value < 0.10)
sum(q.value < 0.30)
sum(q.value < 0.50)
sum(q.value < 0.70)
解析 | 発現変動 | 5群間 | 対応なし | 複製あり | TCC(Sun_2013)
TCCを用いたやり方を示します。
内部的にiDEGES/edgeR(Sun_2013)正規化を実行したのち、
edgeRパッケージ中のexact testで発現変動遺伝子(Differentially Expressed Genes; DEGs)検出を行っています。
TCC原著論文中のiDEGES/edgeR-edgeRという解析パイプラインに相当します。
TCC原著論文(Sun et al., BMC Bioinformatics, 2013)では
多群間複製ありデータ用の推奨パイプラインを示していませんでしたが、多群間比較用の推奨ガイドライン提唱論文
(Tang et al., BMC Bioinformatics, 2015)
で推奨しているパイプライン"EEE-E"が"iDEGES/edgeR-edgeR"と同じものです。この2つの論文を引用し、安心してご利用ください。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
シミュレーションデータ(G1群3サンプル vs. G2群3サンプル vs. G3群3サンプル vs. G4群3サンプル vs. G5群3サンプル)です。
gene_1〜gene_2000までがDEG (gene_1〜gene_1000がG1群で5倍高発現、gene_1001〜gene_1400がG2群で10倍高発現、gene_1401〜gene_1700がG3群で8倍高発現、
gene_1701〜gene_1900がG4群で12倍高発現、gene_1901〜gene_2000がG5群で7倍高発現)、gene_2001〜gene_10000までがnon-DEGであることが既知です。
in_f <- "data_hypodata_3vs3vs3vs3vs3.txt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_G4 <- 3
param_G5 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1,param_G1), rep(2,param_G2), rep(3,param_G3), rep(4,param_G4), rep(5,param_G5))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題1と基本的に同じですが、生のカウントデータではなく正規化後のデータを出力させています。
in_f <- "data_hypodata_3vs3vs3vs3vs3.txt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_G3 <- 3
param_G4 <- 3
param_G5 <- 3
param_FDR <- 0.05
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1,param_G1), rep(2,param_G2), rep(3,param_G3), rep(4,param_G4), rep(5,param_G5))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
tmp <- cbind(rownames(tcc$count), normalized, result)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
- 多群間比較用の推奨ガイドライン提唱論文:Tang et al., BMC Bioinformatics, 2015
- TCC:Sun et al., BMC Bioinformatics, 2013
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- edgeR中のGLM論文:McCarthy et al., Nucleic Acids Res., 2012
解析 | 発現変動 | scRNA-seq | について
single-cell RNA-seq用のプログラムたちです。
通常のRNA-seq用(bulk RNA-seq用)については、「解析 | 発現変動 | RNA-seq | について」をご覧ください。
Soneson and Robinson, Nat Methods, 2018は、
「scRNA-seqに特化した方法よりもbulk RNA-seq用に開発された方法が劣ることはない」といった結論を述べています。
Andrews and Hemberg, F1000Res., 2018は、
dropout問題に起因するゼロカウントの補正アルゴリズムの比較がメインです。
が、この中で発現変動解析の評価も行っており、何もしないデータ(unimputed data)のほうが発現変動解析の場合はよいと述べています(page 8のFigure 2やpage 9の左のほうの記述)。
この主張は、Luecken and Theis, Mol Syst Biol., 2019中の記述とも合致します(page 10のTable 1やpage 11の左中あたり)。
TN testはLuecken and Theis, Mol Syst Biol., 2019のpage 14の左下あたりで言及されています。
Van den Berge et al., Genome Biol., 2018でも、
ZINB-WaVE法(Risso et al., Nat Commun., 2018)
によって得られたgene weights情報とbulk RNA-seq用に開発された方法(edgeR or DESEq2)を組み合わせたやり方がよいということが述べられています
(Luecken and Theis, Mol Syst Biol., 2019のpage 17の左中央あたり)。
このやり方は、weighted bulk DE testingとも表現されています。
解析 | 発現変動 | 時系列 | について
時系列(time-series)解析用のものたちです。
R用:
- Rコード:Nascimento et al., Bioinformatics, 2012
- maSigPro:Nueda et al., Bioinformatics, 2014
- EBSeqHMM (geneとisoformレベル):Leng et al., Bioinformatics, 2015
- FunPat(発現変動、クラスタリング、アノテーションまでだが登録が必要?!):Sanavia et al., BMC Genomics, 2015
- deGPS:Chu et al., BMC Genomics, 2015
- timeSeq(NBMMというモデル):Sun et al., BMC Bioinformatics, 2016
- derfinder:Collado-Torres et al., Nucleic Acids Res., 2016
- ImpulseDE:Sander et al., Bioinformatics, 2017
- tcgsaseq(時系列の遺伝子セット解析):Agniel et al., Biostatistics, 2017
- TSIS(significant transcript isoform switches同定用):Guo et al., Bioinformatics, 2017
- Trendy(うまくパターンの同定を行えそう):Bacher et al., BMC Bioinformatics, 2018
解析 | 発現変動 | 時系列 | maSigPro(Nueda_2014)
maSigProパッケージを用いた時系列データ解析(西岡輔 氏提供情報)を示します。
maSigPro ver. 1.40.0のマニュアルによると、正規化済みのデータを入力とする必要があるようです。
実験デザインの記述などについては、(Rで)マイクロアレイデータ解析でも
maSigProを用いたアレイデータ用の時系列解析も記載しているのでそちらも参考にしてください。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. maSigProパッケージ付属のデータセットの場合:
2群間比較(G1群 vs. G2群)で、6 time points (T1, T2, ..., T6)の正規化済みのデータセット(100 genes ×36 samples)です。
time pointごとに3 replicatesあるので、2群×6time points×3 replicates = 36です。
データはNBdataとして、そして実験デザイン情報はNBdesignとして提供されているので、それを読み込んで実行します。
p.vector関数実行時にcounts=TRUEとしている場合は、NBモデルをしていることに相当します。
最初の20 genes (Gene1, Gene2, ..., Gene20)が経時変化していることが既知なので、これらが検出できれば正解です。
out_f <- "hoge1.txt"
param_FDR <- 0.05
library(maSigPro)
data(NBdata)
data(NBdesign)
data <- NBdata
edesign <- NBdesign
dim(data)
colnames(data)
design <- make.design.matrix(edesign, degree=2,
time.col=1,
repl.col=2,
group.cols=c(3:ncol(edesign)))
design
fit <- p.vector(data, design, counts=TRUE, Q=param_FDR)
fit$i
fit$BH.alfa
fit$SELEC
hoge <- T.fit(fit, alfa=param_FDR)
hoge$sol
out <- get.siggenes(hoge, rsq=0.7, vars="groups")
out$summary
tmp <- cbind(rownames(hoge$sol), hoge$sol)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 発現変動 | 時系列 | Bayesian model-based clustering (Nascimento_2012)
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
選択的スプライシング(alternative splicing; AS)とは、ある共通の前駆体mRNAからエクソン(exon)のextending, shortening, skipping, includingやイントロン(intron)
配列の保持(retaining)によって異なるmRNAバリアントを作り出すメカニズムのことを指します。
これらのイベントはalternative exon events (AEEs)と総称されるようです。
ちなみに全ヒト遺伝子の75-92%が複数のアイソフォーム(multiple isoforms)となりうるという報告もあるようです
(言い換えると8-25%がsingle isoform genesだということ)。
複数のサンプル間で発現に違いのあるエクソン(Differential Exon Usage; DEU)を同定するためのプログラムです。
DEU以外にdifferential isoform usage用の方法が含まれます。
R用:
- Solas(Windows版はなし;2010年以降アップデートなし):Richard et al., Nucleic Acids Res., 2010
- DEXSeq:Anders et al., Genome Res., 2012
- EBSeq:Leng et al., Bioinformatics, 2013
- SplicingCompass(Windows版なし):Aschoff et al., Bioinformatics, 2013
- NPEBseq:Bi et al., BMC Bioinformatics, 2013
- rSeqDiff(Windowsありだが、通常の手順とは異なる):Shi and Jiang, PLoS One, 2013
- DER Finder:Frazee et al., Biostatistics, 2014
- IUTA:Niu et al., BMC Genomics, 2014
- rSeqNP:Shi et al., Bioinformatics, 2015
- MetaDiff:Jia et al., BMC Bioinformatics, 2015
- TSIS(significant transcript isoform switches同定用):Guo et al., Bioinformatics, 2017
- PennDiff:Hu et al., Bioinformatics, 2018
DEXSeqパッケージを用いてサンプル間で発現に違いのあるエクソン(exon)を同定するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | 機能解析 | について
多少間違えているかもしれませんが、とりあえず2018年6月現在の私の理解に基づいて、全貌をざっくりと書きます。
機能解析という項目ですが、実質的にはGene Set Enrichment Analysis (GSEA)を意識した内容です。GSEAは、発現変動解析の枠組みに属するものです。
通常、発現変動解析は発現変動遺伝子(DEG)を個別に検出することを目的として行っています。
そして、その後の解析の多くは、「DEGとして検出されたもの(or 発現変動上位遺伝子群)の中に、何か特定の機能と関連したものが多く濃縮(Enrich)されているかどうか」を調べるというものでした。
例えば、「細胞周期(Cell Cycle)に関連する遺伝子群がDEGの中に多く含まれているかどうか」を調べるというものです。
Mootha et al.,2003の論文では、視点を変えた解析を行っています。
特定の機能に属する遺伝子群に関する知識(knowledge)はGene Ontology(GO)やKEGG Pathwayなどで整理されつつあったので、
「比較するグループ間で、例えば細胞周期に関連する遺伝子群のような特定の機能を果たす遺伝子群(Gene Set)が全体として発現変動しているかどうかを調べる」戦略を提唱しています。
そして、このような知識を利用した解析法(knowledge-based analysis)の考え方をbrush upさせたのが、Subramanian et al., 2005の論文のタイトルでもある、
Gene Set Enrichment Analysis (GSEA)です。この方法は、遺伝子セット解析(Gene Set Analysis; GSA)とも総称されますが、事実上GSEAの考え方そのものを指します。
このような発現変動に関連した機能解析を行う際に、遺伝子セットとして遺伝子オントロジー(GO)の情報を用いる場合はGO解析になり、
遺伝子セットとしてKEGG PathwayやReactomeの情報を用いる場合はパスウェイ解析になります。GSEAが爆発的に流行ったのは、以下に示すような様々な要因が重なったためと考えられます:
- GOやKEGGなど知識の整備が進んでいた(時代背景)
- マイクロアレイもコストが下がり流行っていた(時代背景)
- それらをうまく利用して、従来の機能解析を「知識ベースの発現変動解析」に切り替えた(クールな発想の転換)
- エンドユーザが使いやすいようにMolecular Signatures Database (MSigDB)上で
遺伝子セット解析のための基盤情報を提供(丁寧なアフターフォロー)
MSigDBは、様々な遺伝子セットの情報を含むデータベースです。
GSEAのサイトの右上にある図中で、Gene set Databaseと書かれているものに相当します。
ユーザは、MSigDBから自分が調べたい遺伝子セット情報を含むGMTファイル(.gmt)を予めダウンロードしておく必要があります。
従って、入力ファイルとして必要なものは2種類(マイクロアレイやRNA-seqで得られた発現行列のファイルと.gmtファイル)になります。
これらを入力として、GSEAプログラムそのものや、その後提案された様々な遺伝子セット解析用プログラムを実行するのです。
発現変動に関連した機能解析用プログラムの中には、パスウェイ解析に特化したもの、GO解析もできるもの、遺伝子セット解析全般ができるものなどいろいろあります。
現実問題として、エンドユーザが特に手元にあるRNA-seqの発現データを用いてプログラムを実行する障壁は非常に高いです。
理由は、発現情報ファイル中のfeature IDと.gmtファイル中のIDとの対応付けを行う部分が厄介だからです。
featureという曖昧な用語を用いているのは、発現行列の各行が仮にgeneを指し示すIDに限定されていたとしても、
Ensembl gene ID、Entrez gene ID、gene symbolsなどが現実にあり得ます。また、
exonやtranscriptを指し示すIDかもしれませんし、マイクロアレイデータの場合は各メーカーによって異なる独自のID (例えばAffymetrixのID)になります。
それゆえ、featureという曖昧な表現がよく使われるのです。
現在MSigDBでは、
Entrez gene ID(ファイル名の最後のほうが.entrez.gmt)とgene symbols(ファイル名の最後のほうが.symbols.gmt)
の2種類が提供されています。それゆえ、手元の発現データファイル中のfeature IDがもしEntrez gene IDなら、.entrez.gmtを利用することになります。
feature IDがもしEntrez gene ID以外なら、(多くの場合はgene symbolsとの対応付けは行える状況にあるので)予めfeature IDをgene symbolsにどうにかして変換してから、
.symbols.gmtを利用してプログラムを実行することになります。
但し、発現行列データ側の前処理として、同一feature IDsの重複除去を行っておく必要もあります。
有意な発現変動遺伝子セットを検出する際に、複数個存在する同一feature IDsの情報が過大評価されないようにするのが目的です。
これも、実際に重複除去をやろうとすると色々と厄介です。例えば、発現変動遺伝子セット解析用パッケージのGSVA
は、発現行列データの格納形式としてExpressionSetオブジェクトを利用しています。そして、
前処理として重複除去を行う際にgenefilter
パッケージ内のnsFilter関数(入力がExpressionSetオブジェクト)を利用しています。
ExpressionSetオブジェクトは、特にユーザに意識させることなく(Rで)マイクロアレイデータ解析上でも使っていましたが、
このようなデータ形式を取り扱うスキルもきっちり身につけていく必要があります。尚、
マイクロアレイデータの頃はExpressionSetオブジェクトがよく使われていましたが、RNA-seqカウントデータの現在は
SummarizedExperimentやRangedSummarizedExperimentがよく使われます。
- Gene Set Enrichment Analysis (GSEA):Mootha et al., Nat Genet., 2003
- Gene Set Enrichment Analysis (GSEA):Subramanian et al., PNAS, 2005
- Molecular Signatures Database (MSigDB):Subramanian et al., PNAS, 2005
- Molecular Signatures Database (MSigDB):Liberzon et al., Bioinformatics, 2011
- Gene Ontology (GO):Ashburner et al., Nat Genet., 2000
- Gene Ontology (GO):The Gene Ontology Consortium., Nucleic Acids Res., 2017
- Kyoto Encyclopedia of Genes and Genomes (KEGG):Goto et al., Pac Symp Biocomput., 1997
- Kyoto Encyclopedia of Genes and Genomes (KEGG):Kanehisa et al., Nucleic Acids Res., 2017
- Reactome:Joshi-Tope et al., Nucleic Acids Res., 2005
- Reactome:Fabregat et al., Nucleic Acids Res., 2018
- GSVA:Hänzelmann et al., BMC Bioinformatics, 2013
- genefilter
解析 | 機能解析 | GMTファイル取得 | について
Gene Set Enrichment Analysis (GSEA)に代表される遺伝子セット解析を行うためには、
(発現情報と)遺伝子セット情報が必要です。遺伝子セット情報の取得場所として最も有名なのは、
Molecular Signatures Database (MSigDB)であり、Gene Matrix Transposed (GMT)形式で提供されています。
遺伝子セット情報は、MSigDBを含む以下からも提供されています:
- Gene Set Enrichment Analysis (GSEA):Mootha et al., Nat Genet., 2003
- Gene Set Enrichment Analysis (GSEA):Subramanian et al., PNAS, 2005
- Molecular Signatures Database (MSigDB):Subramanian et al., PNAS, 2005
- Molecular Signatures Database (MSigDB):Liberzon et al., Bioinformatics, 2011
- EGSEAdata:Alhamdoosh et al., F1000Res., 2017
- GeneSetDB:Araki et al., FEBS Open Bio., 2012
- Enrichr(web server):Kuleshov et al., Nucleic Acids Res., 2016
解析 | 機能解析 | GMTファイル取得 | EGSEAdata(Alhamdoosh_2017)
EGSEAdataパッケージからも遺伝子セット情報を取得可能です。
中身はヒトとマウスのMSigDBとGeneSetDBのようです。
GMTファイル取得、と言っていいのかどうかまでは未検証です。
解析 | 機能解析 | GMTファイル取得 | GeneSetDB(Araki_2012)
GeneSetDBからもGMT形式ファイルを取得可能です。
拡張子は.gmtにはなっていませんが、中身は間違いなくGMT形式になっています。GeneSetDB is freely available for academic purposes. だそうです。
解析 | 機能解析 | GMTファイル取得 | MSigDB(Subramanian_2005)
MSigDBから遺伝子セット情報を含むGMTファイル(.gmt)を取得するやり方を示します。2022年5月現在は、MSigDB version 7.5.1です。License Agreement for MSigDB v6.0 and aboveにもありますが、MSigDB v6.0以降、提供データが Creative Commons Attribution 4.0 International Licenseになったようです(KEGG、BioCarta、AAAS/STKE Cell Signaling Database dataの3種類を除く)。
MSigDB v7.5.1では、以下に示す9個の主要なコレクション(9 major collections)が提供されています。ときどきMSigDBからgmtファイルをダウンロードできない事態に遭遇しますので、このサイト上でも提供可能なものはダウンロードできる状態にしています。
但し、このサイト上からダウロードした場合は、registerし、MSigDBに対して仁義を果たして下さい。MSigDBがfunding agenciesに利用者情報を報告するために必要です。
また、MSigDBの論文だけでなく、例えば 1番目のコレクションであるH hallmark gene setsを利用する場合は
Liberzon et al., 2015を、そして
2番目のコレクションであるC2(curated gene sets)に含まれるCP:Reactomeのgene setsを利用する場合は
Joshi-Tope et al., 2005などを適切に引用しましょう。
- H: hallmark gene sets(50 gene sets)
- C1: positional gene sets(299 gene sets)
- C2: curated gene sets(6366 gene sets)
- gene symbols(c2.all.v7.5.1.symbols.gmt)
- entrez genes ids(c2.all.v7.5.1.entrez.gmt)
- CGP: chemical and genetic perturbations(3384 gene sets)
- CP: Canonical pathways(2982 gene sets)
- gene symbols(c2.cp.v7.5.1.symbols.gmt)
- entrez genes ids(c2.cp.v7.5.1.entrez.gmt)
- CP:BIOCARTA: BioCarta gene sets(292 gene sets)
- gene symbols(c2.cp.biocarta.v7.5.1.symbols.gmt)
- entrez genes ids(c2.cp.biocarta.v7.5.1.entrez.gmt)
- CP:KEGG: KEGG gene sets(186 gene sets)
- gene symbols(c2.cp.kegg.v7.5.1.symbols.gmt)
- entrez genes ids(c2.cp.kegg.v7.5.1.entrez.gmt)
- CP:PID: PID gene sets(196 gene sets)
- gene symbols(c2.cp.pid.v7.5.1.symbols.gmt)
- entrez genes ids(c2.cp.pid.v7.5.1.entrez.gmt)
- CP:REACTOME: Reactome gene sets(1615 gene sets)
- CP:WikiPathways: Reactome gene sets(664 gene sets)
- C3: regulatory target gene sets(3726 gene sets)
- C4: computational gene sets(858 gene sets)
- C5: ontology gene sets(15473 gene sets)
- gene symbols(c5.all.v7.5.1.symbols.gmt)
- entrez genes ids(c5.all.v7.5.1.entrez.gmt)
- GO: Gene Ontology gene sets(10402 gene sets)
- BP: biological process(7658 gene sets)
- CC: cellular component(1006 gene sets)
- MF: molecular function(1738 gene sets)
- HPO: Human Phenotype Ontology(5071 gene sets)
- C6: oncogenic signature gene sets(189 gene sets)
- C7: immunologic signature gene sets(5219 gene sets)
- C8: cell type signature gene sets(700 gene sets)
- Molecular Signatures Database (MSigDB):Subramanian et al., PNAS, 2005
- Molecular Signatures Database (MSigDB):Liberzon et al., Bioinformatics, 2011
- Gene Ontology (GO):Ashburner et al., Nat Genet., 2000
- Gene Ontology (GO):The Gene Ontology Consortium., Nucleic Acids Res., 2017
- Kyoto Encyclopedia of Genes and Genomes (KEGG):Goto et al., Pac Symp Biocomput., 1997
- Kyoto Encyclopedia of Genes and Genomes (KEGG):Kanehisa et al., Nucleic Acids Res., 2017
- H hallmark gene sets:Liberzon et al., Cell Syst., 2015
- Reactome:Joshi-Tope et al., Nucleic Acids Res., 2005
- Reactome:Fabregat et al., Nucleic Acids Res., 2018
- transcription factor targets:Xie et al., Nature, 2005
- cancer modules:Segal et al., Nat Genet., 2004
解析 | 機能解析 | GMTファイル読込 | GSEABase(Morgan_2018)
GSEABaseを用いて.gmtファイルを読み込むやり方を示します。
GeneSetCollectionという形式で情報が格納されています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
基本形です。最後のgenesetオブジェクトの確認で、geneIdTypeのところがNullIdentifierに、
そしてcollectionTypeのところがNullCollectionになっているのが分かります。
in_f <- "c1.all.v7.5.1.symbols.gmt"
library(GSEABase)
geneset <- getGmt(in_f)
geneset
このファイルはgene ID情報がgene symbolsであり、
MSigDBの提供元であるBroad Instituteのコレクションの
C1というカテゴリーに属するものであるという情報を明確に与えています。geneIdTypeとcollectionTypeのところが例題1とは異なっていることがわかります。
in_f <- "c1.all.v7.5.1.symbols.gmt"
library(GSEABase)
geneset <- getGmt(in_f, geneIdType=SymbolIdentifier(),
collectionType=BroadCollection(category="c1"))
geneset
例題2とは入力ファイルが異なります。このファイルはgene ID情報がEntrez gene IDsですので、
その部分のみ例題2とは異なります。
in_f <- "c1.all.v7.5.1.entrez.gmt"
library(GSEABase)
geneset <- getGmt(in_f, geneIdType=EntrezIdentifier(),
collectionType=BroadCollection(category="c1"))
geneset
MSigDBの提供元であるBroad Instituteのコレクションの
C2というカテゴリーに属するものであるという情報を明確に与えています。
in_f <- "c2.cp.reactome.v7.5.1.symbols.gmt"
library(GSEABase)
geneset <- getGmt(in_f, geneIdType=SymbolIdentifier(),
collectionType=BroadCollection(category="c2"))
geneset
MSigDBの提供元であるBroad Instituteのコレクションの
C5というカテゴリーに属するものであるという情報を明確に与えています。
in_f <- "c5.mf.v7.5.1.symbols.gmt"
library(GSEABase)
geneset <- getGmt(in_f, geneIdType=SymbolIdentifier(),
collectionType=BroadCollection(category="c5"))
geneset
- GSEABase:原著論文なし
- Molecular Signatures Database (MSigDB):Subramanian et al., PNAS, 2005
- Molecular Signatures Database (MSigDB):Liberzon et al., Bioinformatics, 2011
- Reactome:Joshi-Tope et al., Nucleic Acids Res., 2005
- Reactome:Fabregat et al., Nucleic Acids Res., 2018
- Gene Ontology (GO):Ashburner et al., Nat Genet., 2000
- Gene Ontology (GO):The Gene Ontology Consortium., Nucleic Acids Res., 2017
解析 | 機能解析 | 遺伝子セット解析 | GSVA(Hänzelmann_2013)
GSVAを用いて遺伝子セット解析を行うやり方を示します。
このデータはAbsFilterGSEA
(Yoon et al., PLoS One, 2016)、
およびGSVA
(Hänzelmann et al., BMC Bioinformatics, 2013)中で、
検証用データとして用いられています。具体的には、MSigDBのC1というコレクションに含まれる
2つのsex-specificな遺伝子セット(chryq11とchrxp22)が発現変動しているという結果を得ているようです。従って、
ここではGSVA実行に必要な2つのファイルのうち、gmtファイルをMSigDBから得られた
299 gene setsからなるC1コレクションのgmtファイル(c1.all.v7.5.1.entrez.gmtとc1.all.v7.5.1.symbols.gmt)
を用いて、いくつかの例題を示します。尚、GSVA自体はエンリッチメントスコア(Enrithment Score)をサンプルごとに算出した結果を返すだけなので、
GSVAの実行のみの場合はどのサンプルがどの群に属しているかのグループラベル情報を与える必要はありません。
そのため、GSVA実行結果であるoutオブジェクト(フィルタリング後の遺伝子セット数×サンプル数)を入力として、
とりあえずnon-parametricのシンプルなwilcox.testを実行して得られたp-value情報を取得した結果も出力しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「カウント情報取得 | リアルデータ | SRP001540 | GSVAdata(Hänzelmann_2013)」の例題7を実行して得られた、
11,482 genes × 36 samplesからなる23 females vs. 13 malesの2群間比較用データと同じものです。
同一gene IDsを重複除去した後のデータです。遺伝子セットchryq11のp値が最も低くなっており、妥当ですね。
in_f1 <- "SRP001540_23_13.txt"
in_f2 <- "c1.all.v7.5.1.entrez.gmt"
out_f <- "hoge1.txt"
param_G1 <- 23
param_G2 <- 13
library(GSVA)
library(GSEABase)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
geneset <- getGmt(in_f2, geneIdType=EntrezIdentifier(),
collectionType=BroadCollection(category="c1"))
geneset
data <- as.matrix(data)
out <- gsva(data, geneset,
min.sz=5, max.sz=500, kcdf="Poisson",
mx.diff=T, verbose=F, parallel.sz=1)
dim(out)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
pvalue <- NULL
for(i in 1:nrow(out)){
pvalue <- c(pvalue, wilcox.test(out[i, data.cl==1], out[i, data.cl==2])$p.value)
}
tmp <- cbind(rownames(out), out, pvalue)
tmp <- tmp[order(pvalue),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
「解析 | 前処理 | ID変換 | Ensembl Gene ID --> gene symbols | 基礎」の例題5を実行して得られた、
23,377 gene symbols × 69 samplesからなる40 females vs. 29 malesの2群間比較用データと同じものです。
遺伝子セットchryq11のp値が最も低くなっており、妥当ですね。
in_f1 <- "srp001540_count_symbols.txt"
in_f2 <- "c1.all.v7.5.1.symbols.gmt"
out_f <- "hoge2.txt"
param_G1 <- 40
param_G2 <- 29
library(GSVA)
library(GSEABase)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
geneset <- getGmt(in_f2, geneIdType=SymbolIdentifier(),
collectionType=BroadCollection(category="c1"))
geneset
data <- as.matrix(data)
out <- gsva(data, geneset,
min.sz=5, max.sz=500, kcdf="Poisson",
mx.diff=T, verbose=F, parallel.sz=1)
dim(out)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
pvalue <- NULL
for(i in 1:nrow(out)){
pvalue <- c(pvalue, wilcox.test(out[i, data.cl==1], out[i, data.cl==2])$p.value)
}
tmp <- cbind(rownames(out), out, pvalue)
tmp <- tmp[order(pvalue),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
例題2と基本的に同じですが、gsva実行時に「XXX genes with constant expression values throuhgout the samples」と表示されるXXX genesのリストを抽出するやり方です。
in_f1 <- "srp001540_count_symbols.txt"
in_f2 <- "c1.all.v7.5.1.symbols.gmt"
out_f <- "hoge3.txt"
param_G1 <- 40
param_G2 <- 29
library(GSVA)
library(GSEABase)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
geneset <- getGmt(in_f2, geneIdType=SymbolIdentifier(),
collectionType=BroadCollection(category="c1"))
geneset
data <- as.matrix(data)
out <- gsva(data, geneset,
min.sz=5, max.sz=500, kcdf="Poisson",
mx.diff=T, verbose=F, parallel.sz=1)
dim(out)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
pvalue <- NULL
for(i in 1:nrow(out)){
pvalue <- c(pvalue, wilcox.test(out[i, data.cl==1], out[i, data.cl==2])$p.value)
}
tmp <- cbind(rownames(out), out, pvalue)
tmp <- tmp[order(pvalue),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 機能解析 | 遺伝子オントロジー(GO)解析 | について
RNA-seqなどのタグカウントデータから遺伝子オントロジー(GO)解析を行うためのパッケージもいくつか出ています。
遺伝子セット解析(Gene Set Analysis; GSA)という枠組みではGO解析もパスウェイ解析も同じなので、そちらもチェックするといいかもしれません。
R用:
- GAGE:Luo et al., BMC Bioinformatics, 2009
- goseq:Young et al., Genome Biol., 2010
- GOSemSim:Yu et al., Bioinformatics, 2010
- Rスクリプト:Gao et al., Bioinformatics, 2011
- clusterProfiler:Yu et al., OMICS., 2012
- RamiGO:Schröder et al., Bioinformatics, 2013
- GSVA:Hänzelmann et al., BMC Bioinformatics, 2013
- SeqGSEA(各群5反復以上を要求):Wang et al., Bioinformatics, 2014
- GSAASeqSP:Xiong et al., Sci Rep., 2014
- GOplot(Visualization用):Walter et al., Bioinformatics, 2015
- RNA-Enrich(論文のsuppl):Lee et al., Bioinformatics, 2015
- GOexpress:Rue-Albrecht et al., BMC Bioinformatics, 2016
- rapidGSEA(cudaGSEA and ompGSEA):Hundt et al., BMC Bioinformatics, 2016
- EGSEA:Alhamdoosh et al., Bioinformatics, 2017
- AbsFilterGSEA(small replicates用):Yoon et al., PLoS One, 2016
- GSAR:Rahmatallah et al., BMC Bioinformatics, 2017
- SeqGSA:Ren et al., BioData Min., 2017
- rgsepd:Stamm et al., BMC Bioinformatics, 2019
解析 | 機能解析 | 遺伝子オントロジー(GO)解析 | SeqGSEA (Wang_2014)
SeqGSEAを用いてGO解析を行うやり方を示します。
このパッケージは、exonレベルのカウントデータを入力として、発現変動遺伝子セットに相当する有意に発現変動したGO termsを出力するのが基本ですが、geneレベルのカウントデータを入力として解析することも可能です。
統計的有意性の評価にサンプルラベル情報の並べ替え(permutation)戦略を採用しているため、
各グループあたりの反復数が5以上のデータを想定しているようです(Wang et al., 2014)。また、計算時間が半端なくかかります。
例えば、並べ替え回数がたったの20回でも2時間ちょっとかかります(Panasonic Let's note CF-SX3本郷モデルの場合)のでご注意ください。
推奨は1000回以上と書いてますが、10日ほどかかることになるので個人的にはアリエナイですね...。
SeqGSEAでの機能解析の基本は、exonレベルとgeneレベルの発現変動解析結果を組み合わせてGSEAを行うというものです(Wang and Cairns, 2013)。
SeqGSEA著者たちは、exonレベルの発現変動解析のことをDifferential splicing (DS) analysisと呼んでいて、
おそらくDSGseq (Wang et al., 2013)はSeqGSEA中に組み込まれています。
そしてgeneレベルの発現変動解析をDifferential expression (DE) analysisとして、SeqGSEA中では
DESeqを利用しています。
GSEAに代表される発現変動遺伝子セット解析は、基本的にGSEAの開発者らが作成した様々な遺伝子セット情報を収めた
Molecular Signatures Database (MSigDB)からダウンロードした.gmt形式ファイルを読み込んで解析を行います。
*gmt形式ファイルのダウンロードについては、
「解析 | 機能解析 | GMTファイル取得 | MSigDB(Subramanian_2005)」をご覧下さい。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
パイプライン | ゲノム | 機能解析 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)
のStep2の出力結果ファイルです。SeqGSEA内部でReadCountSetクラスオブジェクトというものを作成する必要がありますが、
これはexonレベルのカウントデータと遺伝子アノテーション情報(Ensembl gene IDおよびexon ID)を対応づけるためのものです。
カウントデータ自体は、ヒトゲノム("hg19")のEnsembl Genes ("ensGene")情報を利用して取得しているので、
アノテーション情報も同じ条件でオンライン上でTxDbオブジェクトとして取得しています(Lawrence et al., 2013)。
以下のスクリプト中の前処理のところでごちゃごちゃと計算しているのは、複数のEnsembl gene IDによって共有されているexon (shared exon)の情報のみ解析から除外しています。
その後、shared exonを除く残りのexonレベルのカウントデータ情報を用いてDifferential splicing (DS) analysisを行い、exonレベルの発現変動解析(Wang et al., Gene, 2013)を行っています。
計算のボトルネックはこの部分です。
次に、exonレベルのカウントデータからgeneレベルのカウントデータを作成したのち、DESeqパッケージを用いて
遺伝子レベルの発現変動解析(Anders and Huber, 2010)を行っています。
SeqGSEA (Wang and Cairns, BMC Bioinformatics)は、これら2つのレベルの発現変動解析結果の情報を統合して
よりよい遺伝子セット解析(Gene Set Enrichment Analysis; GSEA; Subramanian et al., 2005)を行うという手法です。
ヒト遺伝子の90%以上は選択的スプライシングが起こっている(Wang et al., Nature, 2008)ので、
geneレベルのみの発現変動解析結果をもとにするやり方であるGOSeq
(Young et al., Genome Biol., 2010)よりもいいだろう、という思想です。
以下では"c5.bp.v4.0.symbols.gmt"の解析を行っています。
また、並べ替え回数がたったの20回でも2時間ちょっとかかります(Panasonic Let's note CF-SX3本郷モデルの場合)のでご注意ください。
並べ替えを200回行って得られた
srp017142_SeqGSEA_c5bp_exon200.txtでは、FDR < 0.01を満たすGO termは26個であることがわかります。
in_f1 <- "srp017142_count_bowtie2.txt"
in_f2 <- "c5.bp.v4.0.symbols.gmt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_perm <- 20
param1 <- "hg19"
param2 <- "ensGene"
library(SeqGSEA)
library(GenomicFeatures)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
tmp_colname <- colnames(data)
colnames(data) <- c(paste("E", 1:param_G1, sep=""), paste("C", 1:param_G2, sep=""))
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
hoge1 <- exonsBy(txdb, by=c("gene"))
hoge2 <- unlist(hoge1)
hoge2
hoge3 <- table(hoge2$exon_id)
hoge4 <- names(hoge3)[hoge3 == 1]
obj <- is.element(as.character(hoge2$exon_id), hoge4)
exonIDs <- as.character(hoge2$exon_id)[obj]
geneIDs <- names(hoge2)[obj]
data <- data[exonIDs,]
dim(data)
exonIDs <- paste("E", exonIDs, sep="")
RCS <- newReadCountSet(data, exonIDs, geneIDs)
RCS
RCS <- exonTestability(RCS, cutoff = 5)
geneTestable <- geneTestability(RCS)
RCS <- subsetByGenes(RCS, unique(geneID(RCS))[geneTestable])
RCS
time_DS_s <- proc.time()
RCS <- estiExonNBstat(RCS)
RCS <- estiGeneNBstat(RCS)
head(fData(RCS)[, c("exonIDs", "geneIDs", "testable", "NBstat")])
permuteMat <- genpermuteMat(RCS, times=param_perm)
RCS <- DSpermute4GSEA(RCS, permuteMat)
DSscore.normFac <- normFactor(RCS@permute_NBstat_gene)
DSscore <- scoreNormalization(RCS@featureData_gene$NBstat, DSscore.normFac)
DSscore.perm <- scoreNormalization(RCS@permute_NBstat_gene, DSscore.normFac)
RCS <- DSpermutePval(RCS, permuteMat)
head(DSresultGeneTable(RCS))
time_DS_e <- proc.time()
time_DS_e - time_DS_s
time_DE_s <- proc.time()
geneCounts <- getGeneCount(RCS)
head(geneCounts)
DEG <- runDESeq(geneCounts, label(RCS))
DEGres <- DENBStat4GSEA(DEG)
DEpermNBstat <- DENBStatPermut4GSEA(DEG, permuteMat)
DEscore.normFac <- normFactor(DEpermNBstat)
DEscore <- scoreNormalization(DEGres$NBstat, DEscore.normFac)
DEscore.perm <- scoreNormalization(DEpermNBstat, DEscore.normFac)
DEGres <- DEpermutePval(DEGres, DEpermNBstat)
head(DEGres)
time_DE_e <- proc.time()
time_DE_e - time_DE_s
combine <- rankCombine(DEscore, DSscore, DEscore.perm, DSscore.perm, DEweight = 0.3)
gene.score <- combine$geneScore
gene.score.perm <- combine$genePermuteScore
GS <- loadGenesets(in_f2, unique(geneID(RCS)), geneID.type = "ensembl")
GS <- GSEnrichAnalyze(GS, gene.score, gene.score.perm)
#tmp <- GSEAresultTable(GS, GSDesc = TRUE)
tmp <- topGeneSets(GS, n=length(GS@GSNames), sortBy="FDR")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)
のStep2の出力結果ファイルです。
SeqGSEA (Wang et al., 2014)は、
exonレベル(Differential Splicing; DS)とgeneレベル(Differential Expression; DE)の2つの発現変動解析結果を統合して
よりよい遺伝子セット解析(Gene Set Enrichment Analysis; GSEA; Subramanian et al., 2005)を行うという手法ですが、
選択的スプライシング(Alternative Splicing; AS)が少ないあるいはない高等生物以外にも適用可能です。
ここでは、geneレベルの発現変動解析のみに基づくDE-only GSEAのやり方を示します。計算時間のボトルネックになっていたexonレベルの発現変動解析を含まないので高速に計算可能なため、
並べ替え回数を多くすることが可能です(500回で100分程度)。
以下では"c5.bp.v4.0.symbols.gmt"の解析を行っています。並べ替えを500回行って得られた
srp017142_SeqGSEA_c5bp_gene500.txtでは、FDR < 0.05を満たすGO termは10個であることがわかります。
in_f1 <- "srp017142_count_bowtie.txt"
in_f2 <- "c5.bp.v4.0.symbols.gmt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_perm <- 40
library(SeqGSEA)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
tmp_colname <- colnames(data)
colnames(data) <- c(paste("E", 1:param_G1, sep=""), paste("C", 1:param_G2, sep=""))
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
time_s <- proc.time()
DEG <- runDESeq(data, as.factor(data.cl))
DEGres <- DENBStat4GSEA(DEG)
permuteMat <- genpermuteMat(as.factor(data.cl), times=param_perm)
DEpermNBstat <- DENBStatPermut4GSEA(DEG, permuteMat)
DEscore.normFac <- normFactor(DEpermNBstat)
DEscore <- scoreNormalization(DEGres$NBstat, DEscore.normFac)
DEscore.perm <- scoreNormalization(DEpermNBstat, DEscore.normFac)
gene.score <- geneScore(DEscore, DEweight=1)
gene.score.perm <- genePermuteScore(DEscore.perm, DEweight=1)
GS <- loadGenesets(in_f2, rownames(data), geneID.type = "ensembl")
GS <- GSEnrichAnalyze(GS, gene.score, gene.score.perm, weighted.type=1)
time_e <- proc.time()
time_e - time_s
#tmp <- GSEAresultTable(GS, GSDesc = TRUE)
tmp <- topGeneSets(GS, n=length(GS@GSNames), sortBy="FDR")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
- GenomicFeatures:Lawrence et al., PLoS Comput. Biol., 2013
- SeqGSEA(パッケージの論文):Wang et al., Bioinformatics, 2014
- SeqGSEA(の原著論文):Wang and Cairns, BMC Bioinformatics, 2013
- DSGseq:Wang et al., Gene, 2013
- Molecular Signatures Database (MSigDB):Subramanian et al., PNAS, 2005
- DESeq:Anders and Huber, Genome Biol, 2010
解析 | 機能解析 | 遺伝子オントロジー(GO)解析 | GSVA(Hänzelmann_2013)
GSVAを用いて遺伝子オントロジー(GO)解析を行うやり方を示します。
基本的に「解析 | 機能解析 | 遺伝子セット解析 | GSVA(Hänzelmann_2013)」の内容と同じで、gmtファイルが異なるだけです。
ここではGSVA実行に必要な2つのファイルのうち、gmtファイルをMSigDBから得られた7658 gene setsからなるc5.bp.v7.5.1.entrez.gmt
に固定して、いくつかの例題を示します。尚、GSVA自体はエンリッチメントスコア(Enrithment Score)をサンプルごとに算出した結果を返すだけなので、
GSVAの実行のみの場合はどのサンプルがどの群に属しているかのグループラベル情報を与える必要はありません。
そのため、GSVA実行結果であるoutオブジェクト(フィルタリング後の遺伝子セット数×サンプル数)を入力として、
とりあえずnon-parametricのシンプルなwilcox.testを実行して得られたp-value情報を取得した結果も出力しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 11,482 genes × 36 samplesからなるカウントデータファイル(SRP001540_23_13.txt)の場合:
「カウント情報取得 | リアルデータ | SRP001540 | GSVAdata(Hänzelmann_2013)」の例題7を実行して得られた、
23 females vs. 13 malesの2群間比較用データと同じものです。
同一gene IDsを重複除去した後のデータです。3位に遺伝子セットGO_MALE_SEX_DIFFERENTIATIONがきていて妥当ですね。
in_f1 <- "SRP001540_23_13.txt"
in_f2 <- "c5.bp.v7.5.1.entrez.gmt"
out_f <- "hoge1.txt"
param_G1 <- 23
param_G2 <- 13
library(GSVA)
library(GSEABase)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
geneset <- getGmt(in_f2, geneIdType=EntrezIdentifier(),
collectionType=BroadCollection(category="c1"))
geneset
data <- as.matrix(data)
out <- gsva(data, geneset,
min.sz=5, max.sz=500, kcdf="Poisson",
mx.diff=T, verbose=F, parallel.sz=1)
dim(out)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
pvalue <- NULL
for(i in 1:nrow(out)){
pvalue <- c(pvalue, wilcox.test(out[i, data.cl==1], out[i, data.cl==2])$p.value)
}
tmp <- cbind(rownames(out), out, pvalue)
tmp <- tmp[order(pvalue),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 機能解析 | 遺伝子オントロジー(GO)解析 | GOseq (Young_2010)
GOseqを用いてGO解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | 機能解析 | パスウェイ(Pathway)解析 | について
RNA-seqなどのタグカウントデータからパスウェイ(Pathway)解析を行うためのパッケージもいくつか出ています。
入力のカウントデータファイルのgene IDは、Ensembl (Zerbino et al., Nucleic Acids Res., 2018)
が多いようです。
遺伝子セット解析(Gene Set Analysis; GSA)という枠組みではGO解析もパスウェイ解析も同じなので、そちらもチェックするといいかもしれません。
R用:
- KEGGgraph:Zhang et al., Bioinformatics, 2009
- GAGE:Luo et al., BMC Bioinformatics, 2009
- clusterProfiler:Yu et al., OMICS., 2012
- GSVA:Hänzelmann et al., BMC Bioinformatics, 2013
- Pathview:Luo et al., Bioinformatics, 2013
- GSNCA法(GSARで提供されている):Rahmatallah et al., Bioinformatics, 2014
- SeqGSEA:Wang et al., Bioinformatics, 2014
- GSAASeqSP:Xiong et al., Sci Rep., 2014
- seq2pathway:Wang et al., Bioinformatics, 2015
- ToPASeq:Ihnatova and Budinska, BMC Bioinformatics, 2015
- rapidGSEA(cudaGSEA and ompGSEA):Hundt et al., BMC Bioinformatics, 2016
- EGSEA:Alhamdoosh et al., Bioinformatics, 2017
- AbsFilterGSEA(small replicates用):Yoon et al., PLoS One, 2016
- GSAR:Rahmatallah et al., BMC Bioinformatics, 2017
- pathDESeq:Dona et al., Bioinformatics, 2017
- SeqGSA:Ren et al., BioData Min., 2017
- NEArender:Jeggari and Alexeyenko, BMC Bioinformatics, 2017
- tcgsaseq(時系列の遺伝子セット解析):Agniel et al., Biostatistics, 2017
R以外:
- CCS:Schissler et al., Bioinformatics, 2016
- NET-GE(webtool; ヒト専用):Bovo et al., Bioinformatics, 2016
- ContextTRAP(時系列解析用):Lee et al., BMC Bioinformatics, 2016
- MrGSEA(MATLAB):Zyla et al., BMC Bioinformatics, 2017
- NFPscanner(webtool):Xu et al., BMC Bioinformatics, 2017
- EviNet:Jeggari et al., Nucleic Acids Res., 2018
解析 | 機能解析 | パスウェイ(Pathway)解析 | GSAR (Rahmatallah_2017)
まだ途中です。動きません。GSARパッケージを用いた解析のやり方を示します。
GSARは、様々なプログラム群からなるパッケージです。
ここでは、GSARで提供されているGene Sets Net Correlations Analysis法(GSNCA; Rahmatallah et al., 2014)を実行するやり方を示します。
gmtファイルは、MSigDB
(Subramanian et al., PNAS, 2005)から任意のものをダウンロードしてください。
尚、gmtファイルの読み込みにはGSAパッケージ
(Efron and Tibshirani, 2007)中のGSA.read.gmt関数を利用しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. GSARの解析例として示されているPickrell datasetの場合:
tweeDEseqCountDataパッケージから提供されている
pickrellという名前の52,580 genes×69 samplesのデータです。
17 wild-type (WT) samples vs. 33 mutant (MUT) samplesの2群間比較です。
gmtファイルは、MSigDB (ver. 5.2)の
C2: curated gene sets中の
全データファイル(c2.all.v5.2.symbols.gmt)を利用しています。
この中に解析例で取り扱っている遺伝子セット("LU_TUMOR_VASCULATURE_UP")が含まれています。
ここでは、"LU_TUMOR_VASCULATURE_UP"を構成する遺伝子群(29 genes)の中から、
発現データ中に存在するもの(22 genes)に対してGSNCA法を適用した結果のp-value (= 0.02797)を出力するところまでを示します。
p-value = 0.02797という結果は、比較する2群間(WT vs. MUT)でこの遺伝子セットの発現の相関に差がない(帰無仮説)確率が2.797%しかないことを意味します。
1つの判断基準として、(建前上)有意水準α = 0.05を設定していたのなら、帰無仮説が本当は正しいが間違って棄却してしまう確率を5%に設定したことに相当し、
この場合はp-value < 0.05なので帰無仮説を棄却し対立仮説(差がある)を採択することになります。
in_f <- "c2.all.v5.2.symbols.gmt"
param_G1 <- 17
param_G2 <- 33
param <- "LU_TUMOR_VASCULATURE_UP"
library(GSAR)
library(tweeDEseqCountData)
library(GSA)
hoge <- GSA.read.gmt(in_f)
gmt <- hoge$genesets
names(gmt) <- hoge$geneset.names
data(pickrell)
data <- pickrell.eset
rownames(data) <- toupper(rownames(data))
dim(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
set.seed(1053)
hoge <- gmt[[param]]
length(hoge)
obj <- is.element(rownames(data), hoge)
sum(obj)
p.value <- GSNCAtest(data[obj, ], data.cl)
p.value
解析 | 機能解析 | パスウェイ(Pathway)解析 | SeqGSEA (Wang_2014)
SeqGSEAを用いてPathway解析を行うやり方を示します。
このパッケージは、exonレベルのカウントデータを入力として、発現変動遺伝子セットに相当する有意に発現変動したKEGG Pathway名などを出力するのが基本ですが、geneレベルのカウントデータを入力として解析することも可能です。
統計的有意性の評価にサンプルラベル情報の並べ替え(permutation)戦略を採用しているため、
各グループあたりの反復数が5以上のデータを想定しているようです(Wang et al., 2014)。また、計算時間が半端なくかかります。
例えば、並べ替え回数がたったの20回でも2時間ちょっとかかります(Panasonic Let's note CF-SX3本郷モデルの場合)のでご注意ください。
推奨は1000回以上と書いてますが、10日ほどかかることになるので個人的にはアリエナイですね...。
>SeqGSEAでの機能解析の基本は、exonレベルとgeneレベルの発現変動解析結果を組み合わせてGSEAを行うというものです(Wang and Cairns, 2013)。
SeqGSEA著者たちは、exonレベルの発現変動解析のことをDifferential splicing (DS) analysisと呼んでいて、
おそらくDSGseq (Wang et al., 2013)はSeqGSEA中に組み込まれています。
そしてgeneレベルの発現変動解析をDifferential expression (DE) analysisとして、SeqGSEA中では
DESeqを利用しています。
GSEAに代表される発現変動遺伝子セット解析は、基本的にGSEAの開発者らが作成した様々な遺伝子セット情報を収めた
Molecular Signatures Database (MSigDB)からダウンロードした.gmt形式ファイルを読み込んで解析を行います。
*gmt形式ファイルのダウンロード方法は、基本的に以下の通りです:
- Molecular Signatures Database (MSigDB)の
「register」のページで登録し、遺伝子セットをダウンロード可能な状態にする。
- Molecular Signatures Database (MSigDB)の
「Download gene sets」の"Download"のところをクリックし、Loginページで登録したe-mail addressを入力。
- これでMSigDBのダウンロードページに行けるので、
「c2: curated gene sets」の「all canonical pathways」を解析したい場合はc2.cp.v4.0.symbols.gmtファイルをダウンロードしておく。
「c2: curated gene sets」の「BioCarta gene sets」を解析したい場合はc2.cp.biocarta.v4.0.symbols.gmtファイルをダウンロードしておく。
「c2: curated gene sets」の「KEGG gene sets」を解析したい場合はc2.cp.kegg.v4.0.symbols.gmtファイルをダウンロードしておく。
「c2: curated gene sets」の「Reactome gene sets」を解析したい場合はc2.cp.reactome.v4.0.symbols.gmtファイルをダウンロードしておく。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
パイプライン | ゲノム | 機能解析 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)
のStep2の出力結果ファイルです。SeqGSEA内部でReadCountSetクラスオブジェクトというものを作成する必要がありますが、
これはexonレベルのカウントデータと遺伝子アノテーション情報(Ensembl gene IDおよびexon ID)を対応づけるためのものです。
カウントデータ自体は、ヒトゲノム("hg19")のEnsembl Genes ("ensGene")情報を利用して取得しているので、
アノテーション情報も同じ条件でオンライン上でTxDbオブジェクトとして取得しています(Lawrence et al., 2013)。
以下のスクリプト中の前処理のところでごちゃごちゃと計算しているのは、複数のEnsembl gene IDによって共有されているexon (shared exon)の情報のみ解析から除外しています。
その後、shared exonを除く残りのexonレベルのカウントデータ情報を用いてDifferential splicing (DS) analysisを行い、exonレベルの発現変動解析(Wang et al., Gene, 2013)を行っています。
計算のボトルネックはこの部分です。
次に、exonレベルのカウントデータからgeneレベルのカウントデータを作成したのち、DESeqパッケージを用いて
遺伝子レベルの発現変動解析(Anders and Huber, 2010)を行っています。
SeqGSEA (Wang and Cairns, BMC Bioinformatics)は、これら2つのレベルの発現変動解析結果の情報を統合して
よりよい遺伝子セット解析(Gene Set Enrichment Analysis; GSEA; Subramanian et al., 2005)を行うという手法です。
ヒト遺伝子の90%以上は選択的スプライシングが起こっている(Wang et al., Nature, 2008)ので、
geneレベルのみの発現変動解析結果をもとにするやり方であるGOSeq
(Young et al., Genome Biol., 2010)よりもいいだろう、という思想です。
以下では"c2.cp.kegg.v4.0.symbols.gmt"の解析を行っています。
また、並べ替え回数がたったの20回でも2時間ちょっとかかります(Panasonic Let's note CF-SX3本郷モデルの場合)のでご注意ください。
並べ替えを200回行って得られた
srp017142_SeqGSEA_c2cp_exon200.txtでは、FDR < 0.02を満たすパスウェイIDは21個であることがわかります(計算時間は20時間ほどでした)。
in_f1 <- "srp017142_count_bowtie2.txt"
in_f2 <- "c2.cp.kegg.v4.0.symbols.gmt"
out_f <- "hoge1.txt"
param_G1 <- 3
param_G2 <- 3
param_perm <- 20
param1 <- "hg19"
param2 <- "ensGene"
library(SeqGSEA)
library(GenomicFeatures)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
tmp_colname <- colnames(data)
colnames(data) <- c(paste("E", 1:param_G1, sep=""), paste("C", 1:param_G2, sep=""))
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
hoge1 <- exonsBy(txdb, by=c("gene"))
hoge2 <- unlist(hoge1)
hoge2
hoge3 <- table(hoge2$exon_id)
hoge4 <- names(hoge3)[hoge3 == 1]
obj <- is.element(as.character(hoge2$exon_id), hoge4)
exonIDs <- as.character(hoge2$exon_id)[obj]
geneIDs <- names(hoge2)[obj]
data <- data[exonIDs,]
dim(data)
exonIDs <- paste("E", exonIDs, sep="")
RCS <- newReadCountSet(data, exonIDs, geneIDs)
RCS
RCS <- exonTestability(RCS, cutoff = 5)
geneTestable <- geneTestability(RCS)
RCS <- subsetByGenes(RCS, unique(geneID(RCS))[geneTestable])
RCS
time_DS_s <- proc.time()
RCS <- estiExonNBstat(RCS)
RCS <- estiGeneNBstat(RCS)
head(fData(RCS)[, c("exonIDs", "geneIDs", "testable", "NBstat")])
permuteMat <- genpermuteMat(RCS, times=param_perm)
RCS <- DSpermute4GSEA(RCS, permuteMat)
DSscore.normFac <- normFactor(RCS@permute_NBstat_gene)
DSscore <- scoreNormalization(RCS@featureData_gene$NBstat, DSscore.normFac)
DSscore.perm <- scoreNormalization(RCS@permute_NBstat_gene, DSscore.normFac)
RCS <- DSpermutePval(RCS, permuteMat)
head(DSresultGeneTable(RCS))
time_DS_e <- proc.time()
time_DS_e - time_DS_s
time_DE_s <- proc.time()
geneCounts <- getGeneCount(RCS)
head(geneCounts)
DEG <- runDESeq(geneCounts, label(RCS))
DEGres <- DENBStat4GSEA(DEG)
DEpermNBstat <- DENBStatPermut4GSEA(DEG, permuteMat)
DEscore.normFac <- normFactor(DEpermNBstat)
DEscore <- scoreNormalization(DEGres$NBstat, DEscore.normFac)
DEscore.perm <- scoreNormalization(DEpermNBstat, DEscore.normFac)
DEGres <- DEpermutePval(DEGres, DEpermNBstat)
head(DEGres)
time_DE_e <- proc.time()
combine <- rankCombine(DEscore, DSscore, DEscore.perm, DSscore.perm, DEweight = 0.3)
gene.score <- combine$geneScore
gene.score.perm <- combine$genePermuteScore
GS <- loadGenesets(in_f2, unique(geneID(RCS)), geneID.type = "ensembl")
GS <- GSEnrichAnalyze(GS, gene.score, gene.score.perm)
#tmp <- GSEAresultTable(GS, GSDesc = TRUE)
tmp <- topGeneSets(GS, n=length(GS@GSNames), sortBy="FDR")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)
のStep2の出力結果ファイルです。
SeqGSEA (Wang and Cairns, BMC Bioinformatics)は、
exonレベル(Differential Splicing; DS)とgeneレベル(Differential Expression; DE)の2つの発現変動解析結果を統合して
よりよい遺伝子セット解析(Gene Set Enrichment Analysis; GSEA; Subramanian et al., 2005)を行うという手法ですが、
選択的スプライシング(Alternative Splicing; AS)が少ないあるいはない高等生物以外にも適用可能です。
ここでは、geneレベルの発現変動解析のみに基づくDE-only GSEAのやり方を示します。計算時間のボトルネックになっていたexonレベルの発現変動解析を含まないので高速に計算可能なため、
並べ替え回数を多くすることが可能です(500回で100分程度)。
以下では"c2.cp.kegg.v4.0.symbols.gmt"の解析を行っています。並べ替えを500回行って得られた
srp017142_SeqGSEA_c2cpkegg_gene1000.txtでは、FDR < 0.02を満たすPathwayは44個であることがわかります。
in_f1 <- "srp017142_count_bowtie.txt"
in_f2 <- "c2.cp.kegg.v4.0.symbols.gmt"
out_f <- "hoge2.txt"
param_G1 <- 3
param_G2 <- 3
param_perm <- 40
library(SeqGSEA)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
tmp_colname <- colnames(data)
colnames(data) <- c(paste("E", 1:param_G1, sep=""), paste("C", 1:param_G2, sep=""))
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
time_s <- proc.time()
DEG <- runDESeq(data, as.factor(data.cl))
DEGres <- DENBStat4GSEA(DEG)
permuteMat <- genpermuteMat(as.factor(data.cl), times=param_perm)
DEpermNBstat <- DENBStatPermut4GSEA(DEG, permuteMat)
DEscore.normFac <- normFactor(DEpermNBstat)
DEscore <- scoreNormalization(DEGres$NBstat, DEscore.normFac)
DEscore.perm <- scoreNormalization(DEpermNBstat, DEscore.normFac)
gene.score <- geneScore(DEscore, DEweight=1)
gene.score.perm <- genePermuteScore(DEscore.perm, DEweight=1)
GS <- loadGenesets(in_f2, rownames(data), geneID.type = "ensembl")
GS <- GSEnrichAnalyze(GS, gene.score, gene.score.perm, weighted.type=1)
time_e <- proc.time()
time_e - time_s
#tmp <- GSEAresultTable(GS, GSDesc = TRUE)
tmp <- topGeneSets(GS, n=length(GS@GSNames), sortBy="FDR")
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
- GenomicFeatures:Lawrence et al., PLoS Comput. Biol., 2013
- SeqGSEA(パッケージの論文):Wang et al., Bioinformatics, 2014
- SeqGSEA(の原著論文):Wang and Cairns, BMC Bioinformatics, 2013
- DSGseq:Wang et al., Gene, 2013
- Molecular Signatures Database (MSigDB):Subramanian et al., PNAS, 2005
- DESeq:Anders and Huber, Genome Biol, 2010
解析 | 機能解析 | パスウェイ(Pathway)解析 | GSVA(Hänzelmann_2013)
GSVAを用いてパスウェイ(Pathway)解析を行うやり方を示します。
基本的に「解析 | 機能解析 | 遺伝子セット解析 | GSVA(Hänzelmann_2013)」の内容と同じで、gmtファイルが異なるだけです。
ここではGSVA実行に必要な2つのファイルのうち、gmtファイルをMSigDBから得られた1615 gene setsからなるc2.cp.reactome.v7.5.1.entrez.gmt
に固定して、いくつかの例題を示します。尚、GSVA自体はエンリッチメントスコア(Enrithment Score)をサンプルごとに算出した結果を返すだけなので、
GSVAの実行のみの場合はどのサンプルがどの群に属しているかのグループラベル情報を与える必要はありません。
そのため、GSVA実行結果であるoutオブジェクト(フィルタリング後の遺伝子セット数×サンプル数)を入力として、
とりあえずnon-parametricのシンプルなwilcox.testを実行して得られたp-value情報を取得した結果も出力しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 11,482 genes × 36 samplesからなるカウントデータファイル(SRP001540_23_13.txt)の場合:
「カウント情報取得 | リアルデータ | SRP001540 | GSVAdata(Hänzelmann_2013)」の例題7を実行して得られた、
23 females vs. 13 malesの2群間比較用データと同じものです。
同一gene IDsを重複除去した後のデータです。1位の遺伝子セットはREACTOME_NEPHRIN_INTERACTIONSですが、どう解釈していいのかはわかりません。
in_f1 <- "SRP001540_23_13.txt"
in_f2 <- "c2.cp.reactome.v7.5.1.entrez.gmt"
out_f <- "hoge1.txt"
param_G1 <- 23
param_G2 <- 13
library(GSVA)
library(GSEABase)
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
geneset <- getGmt(in_f2, geneIdType=EntrezIdentifier(),
collectionType=BroadCollection(category="c1"))
geneset
data <- as.matrix(data)
out <- gsva(data, geneset,
min.sz=5, max.sz=500, kcdf="Poisson",
mx.diff=T, verbose=F, parallel.sz=1)
dim(out)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
pvalue <- NULL
for(i in 1:nrow(out)){
pvalue <- c(pvalue, wilcox.test(out[i, data.cl==1], out[i, data.cl==2])$p.value)
}
tmp <- cbind(rownames(out), out, pvalue)
tmp <- tmp[order(pvalue),]
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
解析 | 遺伝子制御ネットワーク推定 | について
遺伝子制御ネットワーク(Gene regulatory network; GRN)推定用プログラムたちです。
解析 | 機械学習(分類) | について
未知データが与えられたときに、それがどういう状態かを当てるものたちです(多数の語弊ありw)。
バイオインフォマティクス分野では、癌サンプル群と正常サンプル群のような2群間比較用の数値行列データ(各行が遺伝子、各列がサンプル)
が手元にある状態からスタートします。そしてそのデータのみで全てのことを完結させます。
その理由は、手元のデータのみで完結させることができるからです。
但し!各群のサンプル数が数十程度あるというのが大前提です。例えば、
癌サンプル30例と正常サンプル30例の計60サンプルからなるRNA-seqカウントデータくらいの規模感があるデータじゃないとだめですよ、ということです。
発現変動解析を行う場合は、各群3サンプルで計6サンプルの2群間比較用データを入力とするくらいの感覚です。
しかし、その程度の規模感のデータだと入力として成立しないのでご注意ください。
説明用に、癌サンプルが25例(サンプル名はT1, T2, ..., T25)、正常サンプルが25例(サンプル名はN1, N2, ..., N25)の全部で50サンプルからなるデータを考えます。
まず、手元のデータは、各サンプルの状態(癌 or 正常)が分かっています。
「このサンプルは癌状態のもので、これは正常状態のものだ」ということであり、ラベル情報(label information)
という言い方をします(他には、サンプルラベルとか)。
Step1:方法の選択。機械学習(分類)では、まずどの方法(アルゴリズム)を使うかを決めます。サポートベクターマシン(Support Vector Machine; SVM)とか、
ランダムフォレスト(Random Forest; RF)とか、ニューラルネットワーク(Neural Network; NN)とかいろいろあります。
例えばSVMを使うと決めます。SVMに与える情報には、数値行列データとラベル情報は当然含まれます。
機械学習の最大の目的は、未知サンプルの状態(ラベル情報)を正しく予測するためのモデルを構築することです。
SVMやRFはただの手段であり、予測モデル(判別するための数式という理解でよい)を構築することが重要です。
但し、方法ごとに内部的に用いるパラメータが異なりますが、全てを自動的にやってくれるわけではない点に注意が必要です。
例えば、SVMのときにはどのカーネルを使うか(よく使われるのはRBFカーネル)とか、誤分類をどの程度許容するかというコストパラメータを指定せねばなりません。
このようにヒトが予め適切な値を指定してやらねばならないパラメータのことをハイパーパラメータといいます。
この方法(アルゴリズム)のときはこれらのハイパーパラメータを指定せねばならない、といった情報はネットで取得可能です。
Step2:パラメータチューニング(parameter tuning)。
どのパラメータをどの程度の数値の範囲で何通り試すかというパラメータチューニングも重要です。
このための手段としては、グリッドサーチやクロスバリデーション(交差検証)
グリッドサーチでは、例えばパラメータが2種類あり(パラメータAとB)、パラメータAでは実際の数値として10, 100, 1000を試すとします。
パラメータBでは0.1, 1, 10を試すとします。この場合は3×3 = 9通りの組み合わせを試して最適なパラメータを決めることになります。
Step3:予測と評価。
Step2とリンクしていると思いますが、交差検証ではデータセットをトレーニングセット(training set; 訓練データ)と
テストセット(test set; テストデータ)に分割します。
例えば、計50サンプルのうち、80%をトレーニングセット(T1, T2, ..., T20とN1, N2, ..., N20)、
残りの20%をテストセット(T21, T22, ..., T25とN21, N22, ..., N25)とします。
計9通りのパラメータの組み合わせの1番目を用いて、トレーニングセットに対して予測モデルを構築します。
そして、その予測モデルを用いてテストセットの評価を行います。テストセットは答えが分かっているので、
計10サンプルのうちいくつをうまく当てられたか(予測精度)がわかるのです。次に組み合わせの2番目に対して同様の作業を行い、
予測精度を調べます。こんな感じで3, 4, ..., 9番目まで予測精度を調べてゆけば、どのパラメータを用いればよいかがわかるのです。
もう少し詳細に述べていくと、データセットの分割方法は1通りのみではありません。例えば、データを5分割
(1つ目が1-5、2つ目が6-10、3つ目が11-15、4つ目が16-20、5つ目が21-25番目のサンプル)しておきます。
そうすると、テストセットとして使えるものが5つ分あります。例えば、1つ目の1-5(i.e., T1, T2, ..., T5, N1, N2, ..., N5)をテストセットとし、
残りをトレーニングセットにするのです。こんな感じで結果的に全部のデータをテストセットにしていろいろ予測精度を調べることで、
パラメータチューニングの精度を上げていくことができます。
さらにもう少し詳細に述べていくと、実はこのような交差検証のチューニング手段で得られたパラメータは、
結局のところデータセット内の全ての情報を使っていることになります。それゆえ、結果的に全てのデータを用いて得られた予測精度通りに、
本当の未知のサンプル群に対しても予測できるのかは正当に評価できていないことになります。このあたりが汎化性能(はんかせいのう)
という議論に相当します。それゆえ、例えば、テストセットは本当にテストセットとしてしか使わない、つまりパラメータチューニング用には一切使わないようにするやり方もあります。
それは、例えば、計50サンプルのうち、80%をトレーニングセット(T1, T2, ..., T20とN1, N2, ..., N20)、
残りの20%をテストセット(T21, T22, ..., T25とN21, N22, ..., N25)とした場合に、計40サンプルのトレーニングセットの中で、さらに交差検証のようなことをやって、
パラメータチューニングをやるのです。例えば、計40サンプルのうち、30サンプル(T1, T2, ..., T15とN1, N2, ..., N15)をトレーニングセット内のトレーニングセットとして用い、
トレーニングセット内の残りの10サンプル(T16, T17, T18, T19, T20, N16, N17, N18, N19, N20)を検証セット(validation set)として用いるのです。
次は、本当のテストセット(T21, T22, ..., T25とN21, N22, ..., N25)以外の別の10サンプルを検証セット(例えばT11-T15とN11-N15)とし、
残り(T1-T10とT16-T20、およびN1-N10とN16-N20)をトレーニングセット内のトレーニングセットとして予測モデルを構築するのです。
つまり、手元のデータセットをトレーニングセット、検証セット、テストセットに3分割するようなイメージでパラメータチューニングを行うことで、
汎化性能を正統に評価することができるのです。
ややこしいですが、直感的に(高精度な予測モデルを構築する上で必須である)パラメータチューニングを行いつつ、
且つ正統に予測モデルの性能評価するにはどうしなければいけないだろうか
という視点で考えながら解釈していくとよいと思います。
以下では、機械学習を行うプログラムを示します。
パッケージの右側に「汎用」と書いてあるものの多くは、生命科学分野(RNA-seq, ChIP-seqなど)に限定しないものです。
R用:
- NBLDA:Dong et al., BMC Bioinformatics, 2016
- gcForest(汎用;Deep Forest):Zhi-Hua Zhou ZH and Feng J., arXiv, 2017
- voomDDA:Zararsiz et al., PeerJ, 2017
- DaMiRseq(主にRNA-seqカウントデータ用;多クラス分類も可能):Chiesa et al., Bioinformatics, 2018
- MLSeq(主にRNA-seqカウントデータ用):Goksuluk et al., Comput Methods Programs Biomed., 2019
- pRoloc(mass spectrometry-based spatial proteomics data用):Crook et al., F1000Res., 2019
- scReClassify(scRNA-seq用):Kim et al., BMC Genomics, 2019
- caret(汎用;MLSeqが内部的に利用)
- keras(汎用;Deep Learning or Deep Neural Network)
- randomForest(ランダムフォレスト)
- ranger(ランダムフォレスト)
- Rborist(ランダムフォレスト)
- tensorflow(Googleの機械学習用ライブラリをR上で利用可能にするもの)
解析 | 機械学習(分類) | 基礎 | MLSeq(Goksuluk_2019)
MLSeqを用いて機械学習(分類)を行うやり方を示します。
ここでは入力ファイルをサンプルデータ51のsample51.txtに限定して、
ステップごとに一つ一つ丁寧に説明していきます。このデータは、
MLSeqパッケージから提供されている
cervical.txtという名前のカウントデータと同じものです。714行×58列からなる数値行列データです(「ヘッダー行」や「行名情報の列」を除く)。
データの原著論文は、Witten et al., 2010です。
子宮頸がん患者29例の正常組織と癌組織のペアサンプルであり、714のmicroRNA (714 miRNAs)の発現を調べたデータです。
(行名情報の列を除く)最初の29列分が正常サンプル(N1, N2, ..., N29)、残りの29列分が癌サンプル(T1, T2, ..., T29)のデータです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
Step 1:入力ファイルの読込みとラベル情報の割当てまで。
最初の29列分が正常組織のデータ、残りの29列分が癌組織のデータ
だと分かっている場合に、以下のように記述します。
読み込んだdataオブジェクトは、714行×58列からなる数値行列データとなっていることが分かります。
MLSeqのBeginner's guide
中の「2 Preparing the input data」(page 4)の作業の一部に相当します。
in_f <- "sample51.txt"
param_G1 <- 29
param_G2 <- 29
library(MLSeq)
library(S4Vectors)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t")
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2))
class <- DataFrame(cond=factor(data.cl))
dim(data)
class
以下の事柄は非常にマニアックですが、ドハマりしたので、備忘録的に書いておきます。
上記のdata.clオブジェクト作成部分に関する補足情報です。
クラスラベル情報の与え方次第で、Step 7の予測モデル構築のところでコケマス。
svmやrfのようなcaret-based classifiersを利用する場合に、特に気をつけてください。
voom-basedやdiscrete classifiersの場合は、様々なクラスラベルの指定法に対応してくれているようです。
以下にうまく動く例を示しておきます。
## やり方1
## Step 7で示した全てのアルゴリズムで正常に動作します。
## MLSeqのBeginner's guideのpage4と同じ記述法です。
data.cl <- c(rep("N", param_G1), rep("T", param_G2))
## やり方2
## Step 7で示した全てのアルゴリズムで正常に動作します。
## アルファベットが異なるだけです。Normal vs. Tumorだからといって、NやTにする必要はありません。
data.cl <- c(rep("A", param_G1), rep("B", param_G2))
## やり方3
## Step 7で示した全てのアルゴリズムで正常に動作します。本項目の例題で使われている表記法です。
data.cl <- c(rep("G1", param_G1), rep("G2", param_G2))
## やり方4
## "svmRadial"や"rf"のようなcaret-based classifiersのときにエラーとなります。
data.cl <- c(rep("1", param_G1), rep("2", param_G2))
## やり方5
## Step 7で示した全てのアルゴリズムでエラーとなります。
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
Step 2:分散の大きい上位100行分のデータのみ抽出するところまで。
元の入力データは714行分ありますが、分類を行う上で重要度の高いものから低いものまで様々です。
直感的には、行全体で分散(バラつき)が小さいものは分類問題への寄与が小さいと判断できます。
それゆえ、ここでは行ごとに分散の値を算出し、分散の大きい上位100行分のデータを利用するようにしています。
尚、行数はmicroRNAの種類数に相当します。しかし、実際には行数が遺伝子数だったり転写物数だったりと様々です。
それゆえ、バイオインフォの世界では、より一般的な表現としてfeature(フィーチャー;特徴)という言葉を用います。
そして、ここの作業は、特徴抽出(feature selection)と呼ばれるものに相当します。
入力データの段階でfeature数が100個程度しかないような場合には、ここのステップは行わないのではと思います。
遺伝子発現データのような、数万の遺伝子数(数万のfeature数)からなるデータを取り扱う場合には、
計算コスト削減を主目的としてfeature selectionを行います。
MLSeqのBeginner's guide
中の「3 Splitting the data」(page 5)の作業の一部に相当します。
ここまでの作業で、分散の大きい上位100行分のデータからなるdata.subというオブジェクトを得ています。
param_nfeat <- 100
vars <- apply(data, 1, var, na.rm=T)
head(vars)
vars.sorted <- sort(vars, decreasing=T)
head(vars.sorted)
features <- names(vars.sorted)[1:param_nfeat]
head(features)
data.sub <- data[features, ]
dim(data.sub)
Step 3:データセットの分割。
テストセット(30%)とトレーニングセットを分割。
58*0.3=17.4となるので、ceiling関数を使って切り上げています。
結果として、18サンプルをテストセットとして使うことになります。
sample関数実行時に非復元抽出を意味するreplace=Tとした理由は、同じサンプルを重複して抽出する事態を避けるためです。
set.seed関数内で指定している2128という数値に特に意味はありません(乱数発生時のタネ番号です)。
整数であれば何でもよいはずですが、ここではMLSeqと同じ乱数を発生させるために、同じタネ番号を指定しています。
「前処理(データの分割)」のところでは、データをトレーニングセット(training set)とテストセット(test set)に分割しています。
MLSeqのBeginner's guide
中の「3 Splitting the data」(page 4-5)では、トレーニングセットとして指定する割合のガイドラインを以下のように定めています:
「ほとんどの場合は70%」、「トータルのサンプル数が200-500程度と十分多ければ、80 or 90%」、「サンプル数が少なければ50%」
「サンプル数が十分多ければトレーニングセットが80-90%」というのは妥当です。
理由は、予測モデルを構築するのに十分なトレーニングセット内のサンプル数があり、
且つテストセットが10-20%しかなくともトータルのサンプル数が多いので、テストセットの総数もある程度稼げるからです。
「サンプル数が少なければ50%」というガイドラインから、このパッケージ開発者が
「テストセットで使うサンプル数をある程度確保するのは重要だ」という思想だと判断できます。
MLSeqのBeginner's guide
の4-5ページ目にかけて、「トータル58サンプルのデータの場合は、トレーニングセットのサンプル数を90%にすることもあるかもしれない。」
と書いています。これはマニュアルにも書いているように、
「テストセットを用いた評価よりも、より高精度な予測モデルを構築することを重視」
することに相当します。58サンプルのうち、たった58*0.1 = 6サンプルだけしかテストセットとして用いられない場合は、
たった1サンプルの予測ミスが1/6 = 16.6667%分も予測精度に影響を及ぼすことになります。
それゆえ、テストセットの予測精度が悪かった場合、私の思考回路は以下のような感じになります:
「それはたまたま運が悪かっただけ...かな
トレーニングセット(検証セット含む)での精度はそれなりにあったから...まあしょうがないかな。
一応念のため、トレーニングセットのサンプル数を50-60%くらいに減らして、テストセットを増やしてみようかな。」
param_n.tes <- 0.3
set.seed(2128)
n.tes <- ceiling(ncol(data)*param_n.tes)
n.tes
ind <- sample(x=ncol(data), size=n.tes, replace=F)
ind
tmp <- as.matrix(data.sub + 1)
data.tes <- tmp[, ind]
data.tra <- tmp[, -ind]
dim(data.tes)
dim(data.tra)
class.tes <- DataFrame(cond=class[ind, ])
class.tra <- DataFrame(cond=class[-ind, ])
dim(class.tes)
dim(class.tra)
Step 4:カウントデータとクラスラベル情報をDESeqDataSet形式のオブジェクトに変換。
トレーニングセットとテストセットの情報を、それぞれDESeqDataSetという形式のオブジェクトに格納しています。
DESeq2パッケージが提供している格納形式です。
このパッケージを読み込んだのち、DESeqDataSetFromMatrixという関数を用いてDESeqDataSetという形式のオブジェクトに変換しています。
ここまでで、MLSeqのBeginner's guide
中の「3 Splitting the data」(page 4-5あたり)が終了です。
library(DESeq2)
tes.s4 <- DESeqDataSetFromMatrix(
countData = data.tes,
colData = class.tes,
design = formula(~cond))
tra.s4 <- DESeqDataSetFromMatrix(
countData = data.tra,
colData = class.tra,
design = formula(~cond))
tes.s4
tra.s4
Step 5:利用可能な機械学習アルゴリズムを確認。
MLSeqのBeginner's guide
中の「4 Available machine-learning models」(page 5の下のほう)に相当する部分です。
ここでリストアップされているもののどれかを、この後で利用するclassify関数中のmethodオプションのところで指定します。
packageVersion関数実行結果として、MLSeqのバージョンが2.2.1となっているヒトは、
93個のアルゴリズムが利用可能であることがわかります。
availableMethods()
packageVersion("MLSeq")
Step 6:データの正規化や変換と、用いる機械学習アルゴリズムの組み合わせについて正しく理解。
MLSeqのBeginner's guide
のpage 6の「5 Normalization and transformation」に相当する部分です(page 10付近でも説明されています)。
作業は特になく、Step 5で決めたアルゴリズムによって、この後で利用するclassify関数中のオプションを
うまく使い分けないといけないという話です。
例えば、preProcessingオプションは、入力データに対して「正規化(Normalization)と変換(Transformation)」を実行したい場合に利用します。
normalizeオプションは、入力データに対して「正規化(Normalization)」のみ実行したい場合に利用します。
具体的には、以下に示すように「正規化のみ行って、RNA-seqカウントデータに特化した機械学習アルゴリズムを利用したい場合」と
「正規化だけでなく変換まで行って、多くの一般的な機械学習アルゴリズムを利用したい場合」に大別してオプションを使い分けます。
後者の方で指定する一般的な機械学習アルゴリズムは、caret
パッケージで利用できるものに相当します。これは、MLSeq
が内部的にcaretを利用しているからです。
ここをよく理解してからStep 7に進みます。
6-1. RNA-seqカウントデータに特化した機械学習アルゴリズムを利用したい場合。
・normalizeオプション(以下の3つ):
deseq, tmm, and none
データ正規化までしか行わないのがポイントです。
・methodオプション(以下の6つ):
最初の3つは、PLDA, PLDA2, and NBLDA
discrete classifiersというカテゴリに属するものたちです。
原著論文中では、algorithms which are based on discrete distributionsという表現もなされています。
残りの3つは、voomDLDA, voomDQDA, and voomNSC
voom-based classifiersというカテゴリに属するものたちです。
・controlオプション(以下の2つ):
discrete classifiersを指定した場合は、discreteControlを使います。
voom-based classifiersを指定した場合は、voomControlを使います。
6-2.一般的な機械学習アルゴリズムを利用したい場合。
・preProcessingオプション(以下の4つ):
deseq-vst, deseq-rlog, deseq-logcpm, and tmm-logcpm
データ正規化後に変換まで行うのがポイントです。
・methodオプション(沢山):
knn, rf, svmLinear, glmboost, ctree, and so on.
Step 5の「availableMethods()」実行結果として見られたもののうち、上記の6個以外の全て(のはず)です。
caret-based classifiersとかmicroarray-based classifiersというカテゴリに属するものたちです。
・controlオプション(以下の1つのみ):
trainControl
Step 7:予測モデル(分類器)の構築。
MLSeqのBeginner's guide
中の「6 Model building」(page 7)に相当する部分です。classify関数の実行結果として得られるfitというオブジェクトには、
最適な予測モデル(optimal model)やトレーニングセットに対する性能評価結果が含まれています。
いくつか例示します。多くのアルゴリズムは原著論文が存在しますので適切に引用してください。
下記コードで、classify関数実行時に与えているrefオプションは、
どのクラスラベルがつけられた群をリファレンスとして利用するかを宣言するものです。
このデータの場合は正常サンプル群と癌サンプル群のどちらをリファレンスとして利用するかに相当します。
マニュアルのpage 7では"T"となっていますので、
ここではそれに対応した"G2"をparam_refオブジェクトとして与えています。
refオプションのデフォルトはNULLです。結果として最初に出現するラベル情報である"N"(本項では"G1")とはせずに
わざわざ"T"(本項では"G2")ととした開発者の意図は不明です。
また、e1071パッケージのロードも行っています。
これは、MLSeqのBeginner's guide
中には明記されていませんでしたが、実行時にこのパッケージが必要だと言われたのでつけています。
7-1. RNA-seqカウントデータに特化した機械学習アルゴリズムを利用したい場合。
voom-based classifierの1つである、voom-based Nearest Shrunken Centroids (voomNSC)、
およびdeseq正規化を採用する例です。入力データにはtra.s4を与えています。
controlオプションでは、voomControl関数で定義したオプションを指定しているのがわかります。
詳細についてはMLSeqのReference Manual
中のvoomControlの解説ページに書かれています。tuneLengthオプションのデフォルトは10ですが、
ここでは探索されるチューニングパラメータの総数(total number of tuning parameter to be searched)
を20にしているようです。
param_method <- "voomNSC"
param_normalize <- "deseq"
param_ref <- "G2"
library(e1071)
set.seed(2128)
ctrl <- voomControl(tuneLength=20)
fit <- classify(
data = tra.s4,
method = param_method,
normalize = param_normalize,
ref = param_ref,
control = ctrl)
fit
実行結果であるfitは、MLSeqという形式のクラスオブジェクトです。
fit実行結果として、「機械学習アルゴリズムとしてvoomNSCが使われていること、
Accuracyは97.5%、Sensitivityは94.12%、Specificityは100%、Reference ClassはG2」
が表示されていることがわかります。
str(fit)をやると一気に大量の情報が表示されるのでわかりますが、fitが保有する情報は実際にはもっと沢山あります。
例えば、trained(fit)を実行すると、fitオブジェクト内のtrainedModelというスロットに格納されている情報が表示されます。
fitは計100 features×40samplesからなるトレーニングセットから、2つのクラス("G1" or "G2")のどちらに属するかを決めるために構築された予測モデルです。
「threshold, NonZeroFeat., Accuracy」という情報と、20行分の数値が見えます。
これがclassify関数中のcontrolオプションのところで指定したtuneLength=20の値に対応します。
最もよいAccuracy(=0.9750)が得られたthresholdは2.71944であり、そのときに用いられたfeature数は2であることもわかります。
MLSeqの原著論文(Goksuluk et al., 2019)や
MLSeqのBeginner's guide
のpage 7にも書かれていますが、ここで指定したアルゴリズム(voomNSC)は、
入力情報中の100 features全てを使うわけではありません(voomNSC is sparse and uses a subset of features for classification)。
この中から分類に寄与する少数のfeaturesを抽出しています。
今回の結果では、「100個中2個を使ったときに最も高いAccuracyが得られた」と解釈します。
このあたりの議論が、MLSeqの原著論文(Goksuluk et al., 2019)
のTable 3に記載されている、voomNSCのSparsity = 0.022の結果と対応します。
fit
show(fit)
trained(fit)
fit@modelInfo@trainedModel
fit.voomNSC <- fit
上記trained(fit)でみられるthreshold(横軸)とAccuracy(縦軸)のプロットは以下で得られます。
プロットの数はtuneLength=20で指定した数と対応します。
MLSeqのBeginner's guide
のpage 9のFigure 1に相当します。
7-2. RNA-seqカウントデータに特化した機械学習アルゴリズムを利用したい場合。
voom-based classifierの1つである、voom-based diagonal linear discriminant analysis (voomDLDA)、
およびdeseq正規化を採用する例です。
param_method <- "voomDLDA"
param_normalize <- "deseq"
param_ref <- "G2"
library(e1071)
set.seed(2128)
ctrl <- voomControl(tuneLength=20)
fit <- classify(
data = tra.s4,
method = param_method,
normalize = param_normalize,
ref = param_ref,
control = ctrl)
fit
fit実行結果として、「機械学習アルゴリズムとしてvoomDLDAが使われていること、
Accuracyは92.68%、Sensitivityは94.12%、Specificityは91.67%、Reference ClassはG2」が表示されていることがわかります。
7-1で行ったvoomNSCの結果とは、
上記のパフォーマンスの違いだけでなく「trained(fit)」の実行結果の見栄えも異なります。
MLSeqの原著論文(Goksuluk et al., 2019)や
MLSeqのBeginner's guide
のpage 7にも書かれていますが、ここで指定したアルゴリズム(voomDLDA)は、
入力情報中の100 features全てを使います(VoomDLDA and voomDQDA approaches are non-sparse and use all features to classify the
data)。したがって、実質的にtuneLength=20というオプションは機能していないのだろう、ということが予想されます。
fitオブジェクト中で見られるAccuracy=92.68%と、trained(fit)実行結果として見られるAccuracy=93.75%に違いが見られます。
原著論文著者に問い合わせたところ、The accuracy, in fit object, is calculated with the confusion matrix of train dataset.
However, the second one is calculated by the rounded averages of counts in each cross-validation fold. Actually, you can use any of them.
とのことでした(Thanks to Dr. Zararsiz and Dr. GÖKSÜLÜK)。
fit
show(fit)
trained(fit)
fit@modelInfo@trainedModel
fit.voomDLDA <- fit
上記trained(fit)実行結果中にも「There is no tuning parameter for selected method.」とあることから予想できますが、
下記plot(fit)を実行しても何も表示されません。
MLSeqのBeginner's guide
のpage 9のFigure 1に相当します。
7-3. RNA-seqカウントデータに特化した機械学習アルゴリズムを利用したい場合。
discrete classifierの1つである、negative binomial linear discriminant analysis (NBLDA)、
およびdeseq正規化を採用する例です。
controlオプションではdiscreteControlを指定しているのがわかります。
discreteControl内で与えているtuneLength=10は、
10通りのグリッドサーチを行ってパラメータチューニングを行えという指令です。
number=5は、トレーニングセットの40サンプルのデータを5分割せよという命令です。
1分割あたり40/5 = 8サンプルからなるサブセットが得られますので、
それをさらに8サンプルからなるテストセットと32サンプルからなるトレーニングセットにわけています。
「32サンプルからなるトレーニングセットで予測モデルを構築し、8サンプルからなるテストセットで性能評価を行うという作業」を
repeats=9で指定した回数繰り返しているようなイメージでよいです。このあたりはマニュアルでも丁寧には書かれていません。
repeatsやtuneLengthで指定する数値を大きくするほど時間がかかるようなイメージです。
param_method <- "NBLDA"
param_normalize <- "deseq"
param_ref <- "G2"
library(e1071)
set.seed(2128)
ctrl <- discreteControl(
tuneLength=10,
number=5,
repeats=9)
fit <- classify(
data = tra.s4,
method = param_method,
normalize = param_normalize,
ref = param_ref,
control = ctrl)
fit
fit実行結果として、「機械学習アルゴリズムとしてNBLDAが使われていること、
Accuracyは92.5%、Sensitivityは94.12%、Specificityは91.3%、Reference ClassはG2」が表示されていることがわかります。
上記のパフォーマンスは、repeatsで指定する数値次第でコロコロ変わるようです
(実際、MLSeqのReference Manual中の
page 7あたりでNumber of repeats (repeats) might change model accuracies.という記述もあります。)。
MLSeqの原著論文(Goksuluk et al., 2019)
のTable 3に記載されている、NBLDAのSparsityの値がないこと、
そしてtrained(fit)実行結果で確信(There is no tuning parameter for selected method.という記載)できるように、
tuneLength=10は機能していないのだろうと判断できます。
fit
show(fit)
trained(fit)
fit@modelInfo@trainedModel
fit.NBLDA <- fit
上記trained(fit)実行結果中にも「There is no tuning parameter for selected method.」とあることから予想できますが、
下記plot(fit)を実行しても何も表示されません。
MLSeqのBeginner's guide
のpage 9のFigure 1に相当します。
7-4. 一般的な機械学習アルゴリズムを利用したい場合。
caret-based classifierの1つである、Support Vector Machineおよび
deseq-vst前処理(deseq正規化とvst変換)を採用する例です。
ここでは、データに関する事前知識がない場合によく用いられる、
ラジアル基底関数(Radial basis function; RBF)と呼ばれるカーネル (svmRadial)
を用いてSVMを実行します。
また、preProcessingオプションに変更されているので注意してください。
controlオプションでは、trainControl関数で定義したオプションを指定しているのがわかります。
このtrainControl自体は、caretパッケージで提供されているものです。
classProbsオプションのデフォルトはFalseなようですが、ここではTrueにしています。
classProbs=Tとした根拠は、
MLSeqのBeginner's guide
のpage 7でsvmRadialの実行例がそうなっていたからです。
trainControl関数実行時に、「`repeats` has no meaning for this resampling method.」という警告が出ていることから、
repeatsオプションの数値(ここでは3)を変更しても結果が不変であることが予想されます(実際そうなります)。
tuneLength=7は、SVM実行時に指定するハイパーパラメータである
「誤分類をどの程度許容するかというコストパラメータC」の数値を7通り探索せよという指定です。
注意点としては、classify関数内のオプションとして直接tuneLength=7を与えています。
これは、例題7-1や7-2ではvoomControl関数内にtuneLengthの値を指定していたので、同様にtrainControl関数内に与えたらエラーになったからです。
MLSeqのBeginner's guide
のpage 8でも、classify関数内のオプションとして直接tuneLengthを指定しているので、おそらくこれでよいと思います。
param_method <- "svmRadial"
param_preprocess <- "deseq-vst"
param_ref <- "G2"
library(kernlab)
set.seed(2128)
ctrl <- trainControl(number=2,
repeats=3,
classProbs=T)
fit <- classify(
data = tra.s4,
method = param_method,
preProcessing = param_preprocess,
ref = param_ref,
tuneLength=7,
control = ctrl)
fit
fit実行結果として、「機械学習アルゴリズムとしてsvmRadialが使われていること、
Accuracyは100%、Sensitivityは100%、Specificityは100%、Reference ClassはG2」
が表示されていることがわかります。
MLSeqの原著論文(Goksuluk et al., 2019)
のTable 3に記載されている、SVMのSparsityの値がないことから、
このアルゴリズムは、7-1で用いたようなsparse classifierのカテゴリに属するものではないことがわかります。
また、trained(fit)実行結果から「Resampling: Bootstrapped (2 reps) 」という記述を発見できます。
このことから、この2という数値は、trainControl関数中のnumber=2に対応するものだろうと予想できます。
さらに、「Resampling results across tuning parameters:」という記述から、numberオプションで指定する数値がブートストラップのリサンプリング回数
に対応するのだろうと予想できます。この数値を変えることで、Accuracyが変わるのだろうと予想できます。
今回の結果では「sigma = 0.006054987 and C = 0.5」のパラメータを採用したときに最もよいAccuracy = 0.9198718が得られたと解釈します。
fit
show(fit)
trained(fit)
fit@modelInfo@trainedModel
fit.svmRadial <- fit
上記trained(fit)でみられるコストパラメータC(横軸)とAccuracy(縦軸)のプロットは以下で得られます。
プロットの数はtuneLength=7で指定した数と対応します。
MLSeqのBeginner's guide
のpage 9のFigure 1に相当します。
7-5. 一般的な機械学習アルゴリズムを利用したい場合。
caret-based classifierの1つである、Random Forest (method = "rf")、
およびdeseq-vst前処理(deseq正規化とvst変換)を採用する例です。
7-4.で行ったSVMのときとの違いは見た目上ほとんどありませんが、
trainControl関数実行時に、method = "repeatedcv"を明記している点が異なります。
7-4.では明記せずとも正常に動作しましたが、本例題のRandom Forest (rf)
実行時のみ?!、試行錯誤の末、明示する必要があることを見い出しました。
MLSeqのReference Manual
中のpage 7においてRFの実行例は確かにそうなっていました。
MLSeqのBeginner's guide
のpage 7でも、確かにSVM実行時にmethod = "repeatedcv"を明記してあります。
しかし、7-4.でも示してある通り、明記せずとも正常に動作していたので、rf
実行時のエラー解決に苦労したというオチです。
param_method <- "rf"
param_preprocess <- "deseq-vst"
param_ref <- "G2"
library(randomForest)
set.seed(2128)
ctrl <- trainControl(
method = "repeatedcv",
number=5,
repeats=3,
classProbs=T)
fit <- classify(
data = tra.s4,
method = param_method,
preProcessing = param_preprocess,
ref = param_ref,
tuneLength=7,
control = ctrl)
fit
fit実行結果として、「機械学習アルゴリズムとしてrfが使われていること、
Accuracyは95%、Sensitivityは88.24%、Specificityは100%、Reference ClassはG2」
が表示されていることがわかります。
また、trained(fit)実行結果から「Resampling: Cross-Validated (5 fold, repeated 3 times)」
という記述を発見できます。
このことから、この5という数値はtrainControl関数中のnumber=5
に、そして3という数値はrepeats=3
に対応するものだろうと予想できます。
さらに、「Resampling results across tuning parameters:」という記述から、numberとrepeatsオプションで指定する数値を変えることで、
Accuracyが変わるのだろうと予想できます。
今回の結果では「mtry = 18」を採用したときに最もよいAccuracy = 0.9435185が得られたと解釈します。
fit
show(fit)
trained(fit)
fit@modelInfo@trainedModel
fit.rf <- fit
上記trained(fit)でみられる#Randomly Selected Predictors(横軸)とAccuracy(縦軸)のプロットは以下で得られます。
プロットの数はtuneLength=7で指定した数と対応します。
横軸が18のときに縦軸のAccuracyが最大になっていることから、mtryが#Randomly Selected Predictorsに相当するのだろうと解釈できます。
ここまでで得られた情報から、「Step 2のparam_nfeatで指定した100 featuresの中から
ランダムに2, 18, 34, 51, 67, 83, 100個の計7通り抽出して分類を試し、
結果としてランダムに18個を抽出して分類に用いた結果が最もよくAccuracy = 0.9435185であった」と
解釈すればよいのだろうと断定できます(推測のみで確たる証拠はなし)。
MLSeqのBeginner's guide
のpage 9のFigure 1に相当します。
Step 8:テストセットの予測、および予測精度評価。
MLSeqのBeginner's guide
中の「7 Predicting the class labels of test samples」(page 11)に相当する部分です。
Step 7で構築した予測モデル(分類器)を用いて、Step 4で作成しておいた18サンプルからなるテストセット(tes.s4オブジェクト)の予測を実行します。
このテストセットは、Step 1でG1とラベルした6個のNormalサンプル、
およびG2とラベルした12個のTumorサンプルから構成されていることがわかります。
本当はどのサンプルがどの群に属するかわかっているが、それを知らないものとして予測してみるとどうなるかを調べようとしているのです。
以下ではStep7の例題番号と対応させて、構築した予測モデルで予測を行っています。
8-1. 7-1.で構築した予測モデル(fit.voomNSC)を利用する場合。
outオブジェクトが予測結果です。colnames(input)で示されたサンプル名の順番で、予測結果が示されています。
fit <- fit.voomNSC
input <- tes.s4
out <- predict(object=fit, test.data=input)
out
colnames(input)
rownames(colData(input))
実際には、サンプル名情報は本質的ではなく、サンプルに付随するラベル情報のほうが重要です。
それがcolData(input)$condに相当します。予測結果outと見比べて、異なっているものが予測ミスに相当します。
この場合は、計18サンプル中13サンプルの予測に成功していると判断します。
out
class.tes$cond
colData(input)$cond
colData(input)$cond == out
sum(colData(input)$cond == out)
length(out)
outオブジェクトをみれば分かりますが、Levelsに相当するラベル情報の順番、
特に一番最初に現れるものが"G1"になっています。
Step 7では、リファレンスとして用いるクラスラベル情報を"G2"として実行していました。
ここでもこれをリファレンス(基準)として利用すべく変更すべくrelevel関数を用いて"G2"に変更します。
なんでこれをわざわざやる必要があるのかは、私もいまだによくわかりません。
ここは、MLSeqのBeginner's guide
のpage 12の上のほうの作業に相当します。
param_ref <- "G2"
out
out <- relevel(out, ref=param_ref)
out
colData(input)$cond
actual <- relevel(colData(input)$cond, ref=param_ref)
actual
予測結果outと実際のラベル情報actualを用いて混同行列(confusion matrix)を作成します。
まず、table関数を用いて2×2の分割表(contingency table)を作成したのち、
それを入力としてconfusionMatrix関数を用いて混同行列を作成します。
分割表と混同行列は、見かけ上は同じです。しかし、
正式な混同行列の形式にすることによって、AccuracyやSpecificityなどの値が計算できるのです。
「str(output)」としてoutputの中の構造(structure)を事前に眺めていたので、
output$tableやoutput$overallやoutput$byClassで情報抽出ができることを事前に学んでいます。
fit.voomNSCのAccuracyは0.722であることがわかります。
tbl <- table(Predicted=out, Actual=actual)
output <- confusionMatrix(data=tbl, positive=param_ref)
output
output$table
output$overall
output$byClass
output$overall["Accuracy"]
output$overall["Kappa"]
output$byClass["Sensitivity"]
output.voomNSC <- output
8-2. 7-2.で構築した予測モデル(fit.voomDLDA)を利用する場合。
outオブジェクトが予測結果です。colnames(input)で示されたサンプル名の順番で、予測結果が示されています。
fit <- fit.voomDLDA
input <- tes.s4
out <- predict(object=fit, test.data=input)
out
colnames(input)
rownames(colData(input))
実際には、サンプル名情報は本質的ではなく、サンプルに付随するラベル情報のほうが重要です。
それがcolData(input)$condに相当します。予測結果outと見比べて、異なっているものが予測ミスに相当します。
この場合は、計18サンプル中13サンプルの予測に成功していると判断します。
out
class.tes$cond
colData(input)$cond
colData(input)$cond == out
sum(colData(input)$cond == out)
length(out)
outオブジェクトをみれば分かりますが、Levelsに相当するラベル情報の順番、
特に一番最初に現れるものが"G1"になっています。
Step 7では、リファレンスとして用いるクラスラベル情報を"G2"として実行していました。
ここでもこれをリファレンス(基準)として利用すべく変更すべくrelevel関数を用いて"G2"に変更します。
なんでこれをわざわざやる必要があるのかは、私もいまだによくわかりません。
ここは、MLSeqのBeginner's guide
のpage 12の上のほうの作業に相当します。
param_ref <- "G2"
out
out <- relevel(out, ref=param_ref)
out
colData(input)$cond
actual <- relevel(colData(input)$cond, ref=param_ref)
actual
予測結果outと実際のラベル情報actualを用いて混同行列(confusion matrix)を作成します。
まず、table関数を用いて2×2の分割表(contingency table)を作成したのち、
それを入力としてconfusionMatrix関数を用いて混同行列を作成します。
分割表と混同行列は、見かけ上は同じです。しかし、
正式な混同行列の形式にすることによって、AccuracyやSpecificityなどの値が計算できるのです。
「str(output)」としてoutputの中の構造(structure)を事前に眺めていたので、
output$tableやoutput$overallやoutput$byClassで情報抽出ができることを事前に学んでいます。
fit.voomDLDAのAccuracyは0.889であることがわかります。
tbl <- table(Predicted=out, Actual=actual)
output <- confusionMatrix(data=tbl, positive=param_ref)
output
output$table
output$overall
output$byClass
output$overall["Accuracy"]
output$overall["Kappa"]
output$byClass["Sensitivity"]
output.voomDLDA <- output
8-3. 7-3.で構築した予測モデル(fit.NBLDA)を利用する場合。
outオブジェクトが予測結果です。colnames(input)で示されたサンプル名の順番で、予測結果が示されています。
fit <- fit.NBLDA
input <- tes.s4
out <- predict(object=fit, test.data=input)
out
colnames(input)
rownames(colData(input))
実際には、サンプル名情報は本質的ではなく、サンプルに付随するラベル情報のほうが重要です。
それがcolData(input)$condに相当します。予測結果outと見比べて、異なっているものが予測ミスに相当します。
この場合は、計18サンプル中13サンプルの予測に成功していると判断します。
out
class.tes$cond
colData(input)$cond
colData(input)$cond == out
sum(colData(input)$cond == out)
length(out)
outオブジェクトをみれば分かりますが、Levelsに相当するラベル情報の順番、
特に一番最初に現れるものが"G1"になっています。
Step 7では、リファレンスとして用いるクラスラベル情報を"G2"として実行していました。
ここでもこれをリファレンス(基準)として利用すべく変更すべくrelevel関数を用いて"G2"に変更します。
なんでこれをわざわざやる必要があるのかは、私もいまだによくわかりません。
ここは、MLSeqのBeginner's guide
のpage 12の上のほうの作業に相当します。
param_ref <- "G2"
out
out <- relevel(out, ref=param_ref)
out
colData(input)$cond
actual <- relevel(colData(input)$cond, ref=param_ref)
actual
予測結果outと実際のラベル情報actualを用いて混同行列(confusion matrix)を作成します。
まず、table関数を用いて2×2の分割表(contingency table)を作成したのち、
それを入力としてconfusionMatrix関数を用いて混同行列を作成します。
分割表と混同行列は、見かけ上は同じです。しかし、
正式な混同行列の形式にすることによって、AccuracyやSpecificityなどの値が計算できるのです。
「str(output)」としてoutputの中の構造(structure)を事前に眺めていたので、
output$tableやoutput$overallやoutput$byClassで情報抽出ができることを事前に学んでいます。
fit.NBLDAのAccuracyは0.833であることがわかります。
tbl <- table(Predicted=out, Actual=actual)
output <- confusionMatrix(data=tbl, positive=param_ref)
output
output$table
output$overall
output$byClass
output$overall["Accuracy"]
output$overall["Kappa"]
output$byClass["Sensitivity"]
output.NBLDA <- output
8-4. 7-4.で構築した予測モデル(fit.svmRadial)を利用する場合。
outオブジェクトが予測結果です。colnames(input)で示されたサンプル名の順番で、予測結果が示されています。
fit <- fit.svmRadial
input <- tes.s4
out <- predict(object=fit, test.data=input)
out
colnames(input)
rownames(colData(input))
実際には、サンプル名情報は本質的ではなく、サンプルに付随するラベル情報のほうが重要です。
それがcolData(input)$condに相当します。予測結果outと見比べて、異なっているものが予測ミスに相当します。
この場合は、計18サンプル中13サンプルの予測に成功していると判断します。
out
class.tes$cond
colData(input)$cond
colData(input)$cond == out
sum(colData(input)$cond == out)
length(out)
outオブジェクトをみれば分かりますが、Levelsに相当するラベル情報の順番、
特に一番最初に現れるものが"G1"になっています。
Step 7では、リファレンスとして用いるクラスラベル情報を"G2"として実行していました。
ここでもこれをリファレンス(基準)として利用すべく変更すべくrelevel関数を用いて"G2"に変更します。
なんでこれをわざわざやる必要があるのかは、私もいまだによくわかりません。
ここは、MLSeqのBeginner's guide
のpage 12の上のほうの作業に相当します。
param_ref <- "G2"
out
out <- relevel(out, ref=param_ref)
out
colData(input)$cond
actual <- relevel(colData(input)$cond, ref=param_ref)
actual
予測結果outと実際のラベル情報actualを用いて混同行列(confusion matrix)を作成します。
まず、table関数を用いて2×2の分割表(contingency table)を作成したのち、
それを入力としてconfusionMatrix関数を用いて混同行列を作成します。
分割表と混同行列は、見かけ上は同じです。しかし、
正式な混同行列の形式にすることによって、AccuracyやSpecificityなどの値が計算できるのです。
「str(output)」としてoutputの中の構造(structure)を事前に眺めていたので、
output$tableやoutput$overallやoutput$byClassで情報抽出ができることを事前に学んでいます。
fit.svmRadialのAccuracyは0.944であることがわかります。
tbl <- table(Predicted=out, Actual=actual)
output <- confusionMatrix(data=tbl, positive=param_ref)
output
output$table
output$overall
output$byClass
output$overall["Accuracy"]
output$overall["Kappa"]
output$byClass["Sensitivity"]
output.svmRadial <- output
8-5. 7-5.で構築した予測モデル(fit.rf)を利用する場合。
outオブジェクトが予測結果です。colnames(input)で示されたサンプル名の順番で、予測結果が示されています。
fit <- fit.rf
input <- tes.s4
out <- predict(object=fit, test.data=input)
out
colnames(input)
rownames(colData(input))
実際には、サンプル名情報は本質的ではなく、サンプルに付随するラベル情報のほうが重要です。
それがcolData(input)$condに相当します。予測結果outと見比べて、異なっているものが予測ミスに相当します。
この場合は、計18サンプル中13サンプルの予測に成功していると判断します。
out
class.tes$cond
colData(input)$cond
colData(input)$cond == out
sum(colData(input)$cond == out)
length(out)
outオブジェクトをみれば分かりますが、Levelsに相当するラベル情報の順番、
特に一番最初に現れるものが"G1"になっています。
Step 7では、リファレンスとして用いるクラスラベル情報を"G2"として実行していました。
ここでもこれをリファレンス(基準)として利用すべく変更すべくrelevel関数を用いて"G2"に変更します。
なんでこれをわざわざやる必要があるのかは、私もいまだによくわかりません。
ここは、MLSeqのBeginner's guide
のpage 12の上のほうの作業に相当します。
param_ref <- "G2"
out
out <- relevel(out, ref=param_ref)
out
colData(input)$cond
actual <- relevel(colData(input)$cond, ref=param_ref)
actual
予測結果outと実際のラベル情報actualを用いて混同行列(confusion matrix)を作成します。
まず、table関数を用いて2×2の分割表(contingency table)を作成したのち、
それを入力としてconfusionMatrix関数を用いて混同行列を作成します。
分割表と混同行列は、見かけ上は同じです。しかし、
正式な混同行列の形式にすることによって、AccuracyやSpecificityなどの値が計算できるのです。
「str(output)」としてoutputの中の構造(structure)を事前に眺めていたので、
output$tableやoutput$overallやoutput$byClassで情報抽出ができることを事前に学んでいます。
fit.rfのAccuracyは0.833であることがわかります。
tbl <- table(Predicted=out, Actual=actual)
output <- confusionMatrix(data=tbl, positive=param_ref)
output
output$table
output$overall
output$byClass
output$overall["Accuracy"]
output$overall["Kappa"]
output$byClass["Sensitivity"]
output.rf <- output
Step 9:分類への寄与度の高いfeatures (i.e., possible biomarkers)の情報を取得。
MLSeqのBeginner's guide
中の「9 Determining possible biomarkers using sparse classifiers」(page 14)に相当する部分です。
Step 8とは独立に、Step 7で構築した予測モデルの情報から抽出する情報になります。
voomNSCのようなsparse classifiersの結果のみ、有意義な情報が得られます。
理由は、本項では、Step 2のparam_nfeatで指定した、分散の大きい上位100 featuresを利用していますが、
このうち分類への寄与度を算出するclassifiersは一部だからです。
以下のコマンドを実行すると、sparse classifierであるfit.voomNSCのみ、2つのfeaturesが表示されていることがわかります。
selectedGenes(fit.voomNSC)
selectedGenes(fit.voomDLDA)
selectedGenes(fit.NBLDA)
selectedGenes(fit.svmRadial)
selectedGenes(fit.rf)
trained(fit.voomNSC)@finalModel$model$SelectedGenes
trained(fit.voomDLDA)@finalModel$model$SelectedGenes
trained(fit.NBLDA)@finalModel$model$SelectedGenes
trained(fit.svmRadial)@finalModel$model$SelectedGenes
trained(fit.rf)@finalModel$model$SelectedGenes
- MLSeq:Goksuluk et al., Comput Methods Programs Biomed., 2019
- cervicalデータ原著論文Witten et al., BMC Biol., 2010
- DESeq2:Love et al., Genome Biol., 2014
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- voomDDA(voomNSC):Zararsiz et al., PeerJ, 2017
- voomDLDA:Dudoit et al., J American Stat Assoc., 2002
解析 | 菌叢解析 | について
このあたりはほとんどノータッチです。菌叢解析(microbiome analysis)といういわゆるメタゲノム解析用のRパッケージもあるようです。
分類が正しいのかどうかもよくわかりません。
2017年6月に調査した結果をリストアップします。
R用:
- OTUbase:Beck et al., Bioinformatics, 2011
- mcaGUI:Copeland et al., Bioinformatics, 2012
- phyloseq:McMurdie et al., PLoS One, 2013
- metagenomeSeq:Paulson et al., Nat Methods, 2013
- phylogeo:Charlop-Powers et al., Bioinformatics, 2015
解析 | 菌叢解析 | phyloseq(McMurdie_2013)
phyloseqを用いて菌叢解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | エクソーム解析 | について
目的のエクソン領域のみ効率的に濃縮してシークエンスすることで、SNPやインデル(indel)などエクソン(exon)領域中のの変異を検出するものです。
1,000人ゲノムプロジェクト(1000 Genomes Project Consortium, Nature, 2012)の解析などがこの範疇に含まれます。
basic alignerの一つであるBWAでマッピング後にGenome Analysis Toolkit (GATK)という変異検出プログラムを適用する解析パイプラインが王道のようです。
1,000人ゲノムのようなデータが出てきているのでこれらのpopulation (a collection of genomes)に対して効率的にマッピングを行うBWBBLEというプログラムなどが出始めています。
また、exome sequencingとexome microarrayの比較を行った論文(Wang et al., Front Genet., 2013)なども出ています。
2014年7月に調査した結果をリストアップします。
プログラム:
- BWA:Li and Durbin, Bioinformatics, 2009(BWA-shortの論文)
- BWA:Li and Durbin, Bioinformatics, 2010(BWA-SWの論文)
- GATK:McKenna et al., Genome Res., 2010
- Isaac:Raczy et al., Bioinformatics, 2013
- BWBBLE:Huang et al., Bioinformatics, 2013
- BAYSIC:Cantarel et al., BMC Bioinformatics, 2014
解析 | ChIP-seq | について
このあたりはほとんどノータッチです。
R用:
- ChIPsim:Zhang et al., PLoS Comput. Biol., 2008
- PeakSeq法:Rozowsky et al., Nat Biotechnol., 2009
- CSAR:Kaufmann et al., PLoS Biol., 2009
- rMAT:Droit et al., Bioinformatics, 2010
- ChIPpeakAnno:Zhu et al., BMC Bioinformatics, 2010
- PICS:Zhang et al., Biometrics, 2011
- ChIPseqR:Humburg et al., BMC Bioinformatics, 2011
- DiffBind:Ross-Innes et al., Nature, 2012
- dPeak:Chung et al., PLoS Comput Biol., 2013
- MEDIPS:Lienhard et al., Bioinformatics, 2014
- DSS:Feng et al., Nucleic Acids Res., 2014
- methylSig:Park et al., Bioinformatics, 2014
- ChIPseeker:Yu et al., Bioinformatics, 2015
R以外:
- DROMPA:Nakato et al., Genes Cells., 2013
- bwtool:Pohl and Beato, Bioinformatics, 2014
- SraTailor:Oki et al., Genes Cells., 2014
- annoPeak(webtoolもsource codeもあり):Tang et al., Bioinformatics, 2017
- Ritornello:Stanton et al., Nucleic Acids Res., 2017
- TDCA:Myschyshyn et al., BMC Bioinformatics, 2017
- ChIPulate(Python3。シミュレーション解析用):Datta et al., PLoS Comput Biol., 2019
Review、ガイドライン、パイプライン系:
- ガイドライン:Bailey et al., PLoS Comput Biol., 2013
- HiChIP(パイプライン):Yan et al., BMC Bioinformatics, 2014
- Review:Robinson et al., Front Genet., 2014
- Review:Nakato and Shirahige, Brief Bioinform., 2017
- Review/ガイドライン:de Santiago et al., Methods Mol Biol., 2018
解析 | ChIP-seq | DiffBind (Ross-Innes_2012)
DiffBindを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | ChIPseqR (Humburg_2011)
ChIPseqRを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | chipseq
chipseqを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | PICS (Zhang_2011)
PICSを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | ChIPpeakAnno (Zhu_2010)
ChIPpeakAnnoを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | rMAT (Droit_2010)
rMATを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | CSAR (Kaufmann_2009)
CSARを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | ChIPsim (Zhang_2008)
ChIPsimを用いてChIP-seq解析を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | 新規モチーフ | rGADEM(Li_2009)
rGADEMを用いたやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | ChIP-seq | 新規モチーフ | cosmo(Bembom_2007)
cosmoを用いたやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | chromosome conformation capture (3C) | について
"chromosome conformation capture" (3C)は、DNAが細胞中でどのような立体的な配置(染色体の高次構造; chromosome conformation)をとっているか、
またどのような長距離ゲノム間相互作用(long-range genomic interactions)がなされているかを網羅的に調べるための実験技術です(Dekker et al., Science, 2002)。
その後、circular "chromosome conformation capture" (4C; Zhao et al., Nat. Genet., 2006)、
"chromosome conformation capture" carbon copy (5C; Dostie et al., Genome Res., 2006)、
Hi-C (Lieberman-Aiden et al., Science, 2009)、
single-cell Hi-C (Nagano et al., Nature, 2013)などの関連手法が提案されています。
ReviewとしてはDekker et al., Nat. Rev. Genet., 2013などが参考になると思います。
これらのクロマチン間の相互作用(chromatin interaction)を網羅的に検出すべく、NGSと組み合わされた"3C-seq"由来データが出てきています。
Rパッケージは、マップ後のBAM形式ファイルなどを入力として、統計的に有意な相互作用をレポートします。
2017年04月に調査した結果をリストアップします。
R用:
- r3Cseq:Thongjuea et al., Nucleic Acids Res., 2013
- Basic4Cseq:Walter et al., Bioinformatics, 2014
- FourCSeq:Klein et al., Bioinformatics, 2015
- diffHic:Lun et al., BMC Bioinformatics, 2015
- GOTHiC:Mifsud et al., Bioinformatics, 2017
解析 | 3C | r3Cseq (Thongjuea_2013)
r3Cseqを用いたやり方を示します。
BAM形式ファイルを入力として、シス相互作用(cis-interaction; 同じ染色体上の異なる部位同士の相互作用)や
トランス相互作用(trans-interaction; 異なる染色体間の相互作用)を統計解析結果として出力します。
出力は、シンプルテキストファイルとbedGraphファイル(Kent et al., Genome Res., 2002)のようです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | Bisulfite sequencing (BS-seq) | について
Bisulfite sequencing (BS-seq; Bisulfite-seq)は、ゲノムDNAに対してbisulfite (重亜硫酸ナトリウム)処理を行った後にシーケンスを行う実験技術です。
BS-seqは、メチル化状態を検出するためのものです。bisulfite処理すると、メチル化されていないシトシン(C)はウラシル(U)に変換されます。
しかし、メチル化シトシンはUに変換されません。この性質を利用して、bisulfite処理後のDNAを鋳型にしてsequenceすれば、
メチル化シトシンのみCとして読まれ、それ以外はTとして読まれるので、メチル化の有無を区別することができるというものです。
但し、一般にマイクログラム程度のDNA量を必要とするため、一連の処理はPCR増幅のステップを含みます。
その後、アダプター付加(adapter ligation)後にbisulfite処理を行うのではなく、bisulfite処理後にアダプター付加を行うことでPCR増幅のステップを省いた
PBAT法(post-bisulfite adaptor tagging; Miura et al., Nucleic Acids Res., 2012)などの関連手法が開発されているようです。
2016年10月に調査した結果をリストアップします。
R以外:
- QDMR:Zhang et al., Nucleic Acids Res., 2011
- BiQ Analyzer HT:Lutsik et al., Nucleic Acids Res., 2011
- SAAP-RRBS:Sun et al.,Bioinformatics, 2012
- CpG_MPs:Su et al., Nucleic Acids Res., 2013
- DMEAS:He et al., Bioinformatics, 2013
解析 | BS-seq | BiSeq (Hebestreit_2013)
BiSeqを用いたやり方を示します。
Bismark (Krueger et al., Bioinformatics, 2011)というBS-seq用マッピングプログラムの出力ファイルを入力として、
比較するグループ間でメチル化状態の異なる領域(Differentially Methylated Regions; DMRs)を出力します。
一般に、検定に供されるCpGサイトは100万(one million)以上になります。そのため、多重比較補正(multiple testing correction)後には、
ごく一部の明らかに差のある個所しか有意と判定されない傾向にあります(Bock C., Nat Rev Genet., 2012)。
BiSeqでは、階層的な検定手順(a hierarchical testing procedure; Benjamini and Heller, J Am Stat Assoc., 2007)を採用しています。
具体的には、最初のステップとして「領域(regions)」を単位として検定を行った後、検定の結果棄却された領域(rejected regions)内でより詳細な「個所(locations)」の検定を行う戦略を採用しています。
このように階層的に行うことで、検定数(number of hypothesis tests)を減らすことができ、ひいては検出力(statistical power)を上げることができるわけです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | BS-seq | bsseq (Hansen_2012)
bsseqを用いたやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | 制限酵素切断部位(RECS)地図 | REDseq (Zhu_201X)
制限酵素消化(restriction enzyme digestion; RED)でアプローチ可能なところと不可能なところを区別できるとうれしいようです。
この制限酵素認識部位を調べるためのRパッケージだそうです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
解析 | small RNA | segmentSeq (Hardcastle_2012)
segmentSeqパッケージを用いたやり方を示します。
ゲノム上のsmall interfering RNAs (siRNAs)が結合?!する場所を探してくれるようです。
主な特徴としては、従来の発見的な方法(heuristic method)では、siRNAsの複数個所の"loci"としてレポートしてしまっていたものを、
提案手法の経験ベイズ(empirical Bayesian methods)を用いることで必要以上に分割してしまうことなく"locus"としてレポートしてくれるようです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
karyotype (G banding)のプロットができるものたち?!です。
ComplexHeatmapパッケージを用いるやり方を示します。とりあえず項目のみ。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
NeatMapパッケージを用いるやり方を示します。
サンプルごとに色分けしたクラスタリング結果やヒートマップをを作成することができるようです。
クラスタリングしたあとのサブセットを示すときにも便利そう。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
抗がん剤とプラセボ(偽薬)のどちらがより長く生存したかを示すような、生存率曲線(生存曲線)を描くものたちです。
縦軸が累積生存率、横軸が生存時間です。カプランマイヤー(Kaplan-Meier)法を用いて比較する2群の生存率を同時に評価する場合が多い?!ので、
一般のヒトにとっては、「カプランマイヤー」という言葉のほうが馴染みがあると思います。
抗がん剤を使ったかどうかという原因側のデータを説明変数、
結果としてどれだけ長生きできたかという結果側の変数を目的変数と言います。
原因側のデータは、女か男かで調べてもよいですし、病気の種類でもよいです。
「病気A, B, C」のように3種類以上のような場合でも指定可能です。
また、「病気A, B, C」と「男女」を組み合わせて調べることだってできます。ここでは、主にsurvival
パッケージを用いています。Surv関数のところで指定するのは、列名情報です。
「uge ~ ege」のような感じで指定しますが、右側に原因側である説明変数を指定し、
左側に結果側である目的変数を指定します。
survivalパッケージを用いた生存曲線(カプランマイヤー曲線)
の描画を行うやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
MASSパッケージから提供されている
gehanという名前の生存時間解析用データと同じものです。これは(ヘッダー行を除く)42行×4列からなる数値行列データです。
42行の行数は、42人の白血病患者(leukemia patients)数に相当します。
2人ずつのペアになっており、片方には6-mercaptopurine (6-MP)という薬を投与、もう片方にはプラセボ(control)を投与しています。
行列データの各列には以下に示す情報が格納されています:
1列目(列名:pair)は、患者のid情報が示されています。
例えば1-2行目が1番目のペア、3-4行目が2番目のペアだと読み解きます。
2列目(列名:time)は、寛解時間(単位は週)です。MASSのリファレンスマニュアル56ページ目では、
remission time in weeksと書いてあります。大まかには「元気に過ごせた時間」とか「生存時間」のように解釈しちゃって構いません。
3列目(列名:cens)は、打ち切り(censoring)があったかなかったかという 0 or 1の情報からなります。打ち切りがあったら0、なかったら1です。
このデータの場合は、3列目の0が12個、1が30個です。したがって、12人の患者さんのデータが打ち切りのあるデータ(「上完全データ」と呼ぶそうです)、
30人の患者さんのデータが打ち切りのないデータ(「完全データ」と呼ぶそうです。)ということになります。打ち切りデータというのは、
患者さんとの連絡が取れなくなったなど、何らかの理由で患者さんの状況を把握する手段がなくなったデータのことを指します。
観察期間終了まで生存されている患者さんの場合も、「打ち切りありで0」ということになります。
ちなみに、亡くなったという情報が分かっているデータは打ち切りのないデータに相当します。
4列目(列名:treat)には、プラセボ(control)投与群か6-MP投与群かという「どのような処理を行ったかという処理(treatment)情報」が記載されています。
ここでは、入力ファイル中のtreat列を、原因側である説明変数として指定しています。
結果側である目的変数で指定している部分は、「Surv(time=time, event=cens)」
に相当します。イベント(打ち切り)情報であるcens列の情報を加えた、time列の情報を、
Survという関数を用いて生存時間解析用のオブジェクトに変換しています。
抗がん剤(6-MP)投与群のほうがプラセボ(control)投与群よりも時間が長いことが分かります。
in_f <- "sample48.txt"
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate")
survivalパッケージから提供されている
kidneyという名前の生存時間解析用データ(sample49.txt;タブ区切りテキストファイル)です。
これは(ヘッダー行を除く)76行×7列からなる数値行列データです。
これは、ポータブル透析装置(portable dialysis equipment)を使用している腎臓病患者(kidney patients)向けの、
カテーテル(catheter)挿入時点での感染までの再発時間に関するデータです(McGilchrist and Aisbett, Biometrics, 1991)。
カテーテルは感染以外の理由で除去される場合があります。その場合、観察は打ち切られます。
患者1人につき、2つの観察結果(2 observations)があります。
このデータは76行ありますので、76/2 = 38人分の腎臓病患者のデータがあることになります。
このデータは、生存モデルでのランダム効果(フレイル)を説明するためによく使用されているようです。
行列データの各列には以下に示す情報が格納されています:
1列目(列名:id)は、患者のid情報が示されています。
例えば、(ヘッダー行を除く)最初の1-2行がid = 1の最初の患者さん、次の3-4行がid = 2の患者さん、という風に解釈します。
2列目(列名:time)は、時間です(単位不明)。
3列目(列名:status)は、event statusです。
0 or 1ですが、何が0で何が1かは記載されていません。
Rと生存時間分析(2)
によると、「打ち切りは0、その他は1」となっています。
4列目(列名:age)は、年令情報(in years)です。
5列目(列名:sex)は、性別情報です。1が男性、2が女性です。
6列目(列名:disease)は、「disease type」に関する情報が含まれています。
「0=GN, 1=AN, 2=PKD, 3=Other」だそうです。PKDはほぼ間違いなくpolycystic kidney diseaseの略で、多発性囊胞腎です。
ANは、おそらくacute nephritis (急性腎炎)のこと。GNは、glomerular nephritis (糸球体腎炎)の略なんだろうとは思いますが...。
実際には、数値ではなく「GN or AN or PKD or Other」のいずれかが記載されています。
7列目(列名:frail)は、「frailty estimate from original paper」に関する情報が含まれています。
原著論文(McGilchrist and Aisbett, Biometrics, 1991)から推定した
フレイル(frailty;加齢により心身が老い衰えた状態)の度合いを数値で示したもののようです。数値が大きいほど衰えの度合いが高い?!。
ここでは、入力ファイル中のsex列を、原因側である説明変数として指定しています。
結果側である目的変数で指定している部分は、「Surv(time=time, event=status)」
に相当します。イベント(打ち切り)情報であるstatus列の情報を加えた、time列の情報を、
Survという関数を用いて生存時間解析用のオブジェクトに変換しています。
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
例題2と入力ファイルは同じですが、sex列(性別情報)ではなく、
disease列(disease type;病気の種類情報)を、原因側である説明変数として指定しています。
それ以外は同じです。
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
例題3と入力ファイルは同じですが、sex列(性別情報)と
disease列(disease type;病気の種類情報)を、原因側である説明変数として指定しています。
それ以外は同じです。
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
sfオブジェクトの実行結果を眺めても分かりますが、例えば「男でAN(sex=1, disease=AN)」はn=4でevents=3であることがわかります。
これは、データ数は4 (idが16と25のヒト)であり、そのうち打ち切りなしのデータが3つ(status列の1の個数)であることを意味します。
median=17とありますが、これは打ち切りなしデータの3つのtime情報(12, 17, 40)の中央値に相当するのだろうということがわかります。
マニュアルの説明文を読んだだけでは理解できないことが多いですが、こんな感じで元の入力ファイルと突き合わせて確認しながら理解してゆけばよいです。
尚、以下のコードでは「sex+disease」と記述していますが、
「disease+sex」でも構いません。sfオブジェクト内の順番が入れ替わるだけだからです。
in_f <- "sample49.txt"
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
survivalパッケージから提供されている
colonという名前の生存時間解析用データ(sample50.txt;タブ区切りテキストファイル)です。
カプランマイヤー(Kaplan-Meier)法による生存曲線(カプランマイヤー曲線;生存率曲線)作成時の入力ファイルです。
survivalのリファレンスマニュアル23ページ目のcolonの説明部分でも解説されていますが、
これは(ヘッダー行を除く)1858行×16列からなる数値行列データです。
これは、大腸がんに対する術後補助化学療法(adjuvant chemotherapy)の有効性を示したデータです
(Laurie et al., J Clin Oncol., 1989)。
化学療法としては、低毒性のレバミゾール(Levamisole; 線虫駆虫薬の1種)と、
中程度の毒性のフルオロウラシル(fluorouracil; 5-FU)が使われています。
患者は、何もせずに経過観察(Obsavation)のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類に分けられます。
患者1人につき、2つの記録(two records)があります。再発(recurrence)が1で、死亡(death)が2です。
このデータは1858行ありますので、1858/2 = 929人分の大腸がん患者(colon cancer patients)のデータがあることになります。
それぞれのイベントの種類(event type; etype)ごとに、時間(time)の情報があります。
例えば、idが1の患者さんは、再発(etype = 1)までの時間が968 days、死亡(etype = 2)までの時間が1521 daysだったと解釈します。
データ全体を眺める(特にtime列とetype列を見比べる)とわかりますが、死亡までの時間のほうが再発までの時間よりも短いデータはアリエマセン。
「再発までの時間 <= 死亡までの時間」ということになります。
行列データの各列には以下に示す情報が格納されています:
1列目(列名:id)は、患者のid情報が示されています。
例えば、(ヘッダー行を除く)最初の1-2行がid = 1の最初の患者さん、次の3-4行がid = 2の患者さん、という風に解釈します。
2列目(列名:study)は、全て1であり特に意味はありません。
3列目(列名:rx)は、患者に対してどのような処置(Treatment)を行ったのかという情報が含まれています。
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類です。
4列目(列名:sex)は、性別情報です。0が女性、1が男性です。
5列目(列名:age)は、年令情報(in years)です。
6列目(列名:obstruct)は、「obstruction of colon by tumour」に関する情報が含まれています。
おそらく癌によって腸閉塞が行ったかどうかという0 or 1の情報からなるのだろうと思います。この列をざっと眺めると、0のほうが多いこと、
そしてそれほど腸閉塞の頻度は多くないだろうという素人判断から、0が腸閉塞なし、1が腸閉塞ありなのだろうと思います。
7列目(列名:perfor)は、「perforation of colon」に関する情報が含まれています。
おそらく結腸(大腸のこと)に穴(穿孔)があいちゃったかどうかという0 or 1の情報からなるのだろうと思います。この列をざっと眺めると、0のほうが多いこと、
そしてそれほど穿孔の頻度は多くないだろうという素人判断から、0が穿孔なし、1が穿孔ありなのだろうと思います。
8列目(列名:adhere)は、「adherence to nearby organs」に関する情報が含まれています。
近くの臓器への癒着があったかどうかという0 or 1の情報からなるのだろうと思います。この列をざっと眺めると、0のほうが多いこと、
そしてそれほど癒着の頻度は多くないだろうという素人判断から、0が癒着なし、1が癒着ありなのだろうと思います。
9列目(列名:nodes)は、「number of lymph nodes with detectable cancer」に関する情報が含まれています。
リンパ節への転移に関する情報で、転移のあったリンパ節数(多いほどよくない)です。
10列目(列名:status)は、「censoring status」に関する情報が含まれています。
打ち切り(censoring)があったかなかったかという 0 or 1の情報からなります。打ち切りがあったら0、なかったら1です。
打ち切りデータというのは、患者さんとの連絡が取れなくなったなど、何らかの理由で患者さんの状況を把握する手段がなくなったデータのことを指します。
観察期間終了まで生存されている患者さんの場合は、「打ち切りありで0」ということになります。
ちなみに、亡くなったという情報が分かっているデータは打ち切りのないデータに相当します。
11列目(列名:differ)は、「differentiation of tumour」に関する情報が含まれています。
分化度(differentiation)のことですね。「1=well, 2=moderate, 3=poor」です。高分化型が1、中分化型が2、低分化型が3です。
数値が大きいほど悪性度が高いと解釈します。
12列目(列名:extent)は、「Extent of local spread」に関する情報が含まれています。
「腫瘍の局所的拡大の範囲」と解釈すればよいのでしょうか。「1=submucosa, 2=muscle, 3=serosa, 4=contiguous structures)」です。
大腸粘膜(mucosa)の次の層がsubmucosa、その次が筋層(muscle layer)、その次がserosa (serous membrane;漿膜)、
最後にcontiguous structures (直接隣接する組織)となります。数値が大きいほど深層まで達していると解釈できるので、悪性度が高いと解釈します。
13列目(列名:surg)は、「time from surgery to registration」に関する情報が含まれています。
来訪してから手術までの期間であり、「0=short, 1=long」です。
14列目(列名:node4)は、「more than 4 positive lymph nodes」に関する情報が含まれています。
nodes列が4よりも大きいものが1、4以下が0となっているようですね。
15列目(列名:time)は、「days until event or censoring」に関する情報が含まれています。
イベント(再発 or 死亡)または打ち切り(censoring)までの日数です。数値の大きさは、生存時間の長さを表します。
16列目(列名:etype)は、「event type」に関する情報が含まれています。
再発(recurrence)が1で、死亡(death)が2です。
ここでは、入力ファイル中のsex列を、原因側である説明変数として指定しています。
結果側である目的変数で指定している部分は、「Surv(time=time, event=status)」
に相当します。イベント(打ち切り)情報であるstatus列の情報を加えた、time列の情報を、
Survという関数を用いて生存時間解析用のオブジェクトに変換しています。
sfオブジェクトの実行結果より、Timeが2500くらいまでは、890人いる女性(sex=0)のほうが968人いる男性(sex=1)よりも生存時間が短いことがわかります。
in_f <- "sample50.txt"
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
plot(sf, xlab="Time(in days)", ylab="Survival rate")
例題5と入力ファイルは同じですが、sex列(性別情報)ではなく、
node4列を、原因側である説明変数として指定しています。
この列は、転移のあったリンパ節数(多いほどよくない)が、4個以下なら0、4個よりも多ければ1とした情報です。
予想としては、リンパ節転移が少ないほうが生存率が高いはずであり、実際に2つの状態間で明瞭に生存曲線が分離されていることがわかります。
sfオブジェクトの実行結果より、n=1348の転移したリンパ節数が4個以下(node4=0)のヒトの生存時間の中央値はNA、
n=510の転移したリンパ節数が4個よりも多い(node4=1)ヒトの生存時間の中央値は697日であることがわかります。
この段階で、生存曲線の悪いほうがnode4=1のデータであることがわかります。理由は、図の縦軸が0.5のところ
で水平線を引き、曲線との交点から垂直に下した横軸(Time)の値が大体700 days付近であるからです。
もう一つのほうは、縦軸が0.5まで達していないので、medianを計算できないのだろうと解釈できます。
in_f <- "sample50.txt"
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
plot(sf, xlab="Time(in days)", ylab="Survival rate")
例題6と入力ファイルは同じですが、
rx列を、原因側である説明変数として指定しています。
この列は、患者に対してどのような処置(Treatment)を行ったのかという情報が含まれています。
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類です。
1つの群のみ生存率がよいことが分かります。sf実行結果で、median=NAとなっているのは「rx=Lev+5FUの群」であり、
Time = 3000のあたりまで見ても確かに縦軸の生存率(Survival rate)が0.6程度あることが分かります。
原著論文(Laurie et al., J Clin Oncol., 1989)が補助化学療法として5-FU
の効果を主張したものであることからも妥当ですね。
in_f <- "sample50.txt"
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
plot(sf, xlab="Time(in days)", ylab="Survival rate")
ここでは、「1. まずはプロット」の続きとして、縦横のサイズを指定してPNG形式ファイルとして保存するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(sf, xlab="Time(in weeks)", ylab="Survival rate")
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
dev.off()
Timeが2500以降ではっきり差がついていることがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(sf, xlab="Time(in days)", ylab="Survival rate")
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(sf, xlab="Time(in days)", ylab="Survival rate")
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(sf, xlab="Time(in days)", ylab="Survival rate")
dev.off()
この項目では、図の余白を任意に変更します。
「下、左、上、右」の順で余白を指定します。単位は「行」で、0が最も余白が小さく、大体5程度が最大です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate")
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate")
dev.off()
Timeが2500以降ではっきり差がついていることがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate")
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate")
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate")
dev.off()
この項目では、plot関数実行時に、cex.labとcex.axisオプションを追加して、軸ラベルや数値の大きさを変えます。
通常のx倍という指定の仕方をします。
例えば、cex.lab=1.5は軸ラベル名を通常の1.5倍にしてね、ということに相当します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5)
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5)
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5)
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5)
dev.off()
Timeが2500以降ではっきり差がついていることがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5)
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5)
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5)
dev.off()
この項目では、colオプションを追加して色分けします。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue", "pink")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
色の名前を8種類も打つのが面倒なのでデフォルトの色番号(1:black, 2:red, ...)で指定しています。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- 1:8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
dev.off()
Timeが2500くらいまでは、890人いる女性(sex=0)のほうが968人いる男性(sex=1)よりも生存時間が短いことがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
dev.off()
この項目では、grid関数を用いてグリッドを追加します。ltyオプションでは、線分の形式を指定します。
"dotted"(点線)の代わりに、"dashed"(破線)や"solid"(実線)も指定可能です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue", "pink")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
色の名前を8種類も打つのが面倒なのでデフォルトの色番号(1:black, 2:red, ...)で指定しています。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- 1:8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
Timeが2500くらいまでは、890人いる女性(sex=0)のほうが968人いる男性(sex=1)よりも生存時間が短いことがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
この項目では、legend関数を用いて凡例を追加します。legend関数実行時のlty=1は、
線分の形式を実線にするように指定していることに相当します。
lty=2は破線に相当します。他にもいろいろあります。
cex=1.5というのは、線分の大きさの倍率指定です。この場合は通常の1.5倍に相当します。
凡例で指定する文字列の並びは、sfオブジェクトの中身を見ながら、その出現順に記載していきます。
本当はもう少し自動化できるやり方はあるのですが、初心者のうちはその解釈が難解だと思うのでやめてます。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("6-MP", "Control")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Male", "Female")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue", "pink")
param_legend <- c("AN", "GN", "Other", "PKD")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
色の名前を8種類も打つのが面倒なのでデフォルトの色番号(1:black, 2:red, ...)で指定しています。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- 1:8
param_legend <- c("Male_AN", "Male_GN", "Male_Other", "Male_PKD", "Female_AN", "Female_GN", "Female_Other", "Female_PKD")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
Timeが2500くらいまでは、890人いる女性(sex=0)のほうが968人いる男性(sex=1)よりも生存時間が短いことがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Female", "Male")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("node4(less)", "node4(more)")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue")
param_legend <- c("Lev", "Lev+5FU", "Obs")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
この項目では、plot関数実行時にconf.int=Tオプションを付けて、デフォルトの95%信頼区間も描画しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("6-MP", "Control")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Male", "Female")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue", "pink")
param_legend <- c("AN", "GN", "Other", "PKD")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
色の名前を8種類も打つのが面倒なのでデフォルトの色番号(1:black, 2:red, ...)で指定しています。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- 1:8
param_legend <- c("Male_AN", "Male_GN", "Male_Other", "Male_PKD", "Female_AN", "Female_GN", "Female_Other", "Female_PKD")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
Timeが2500くらいまでは、890人いる女性(sex=0)のほうが968人いる男性(sex=1)よりも生存時間が短いことがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Female", "Male")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("node4(less)", "node4(more)")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue")
param_legend <- c("Lev", "Lev+5FU", "Obs")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
この項目では、survfit関数実行時にconf.intオプションを付けて、80%信頼区間も描画しています。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("6-MP", "Control")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Male", "Female")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue", "pink")
param_legend <- c("AN", "GN", "Other", "PKD")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
色の名前を8種類も打つのが面倒なのでデフォルトの色番号(1:black, 2:red, ...)で指定しています。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- 1:8
param_legend <- c("Male_AN", "Male_GN", "Male_Other", "Male_PKD", "Female_AN", "Female_GN", "Female_Other", "Female_PKD")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
Timeが2500くらいまでは、890人いる女性(sex=0)のほうが968人いる男性(sex=1)よりも生存時間が短いことがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Female", "Male")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("node4(less)", "node4(more)")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue")
param_legend <- c("Lev", "Lev+5FU", "Obs")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
この項目では、信頼区間の凡例を追加します。legend関数実行時のオプションに、例えばlty=c(1,2,1,2)があります。
これは凡例の表示順に、線分を「実線(lty=1)・破線(lty=2)・実線(lty=1)・破線(lty=2)」の順で描画してね、という指令です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
in_f <- "sample48.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("6-MP", "Control")
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=cens)~treat
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
summary(sf)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in weeks)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
uge <- paste(100*param_confint, "%信頼区間", sep="")
legend("topright", legend=c("6-MP", uge, "control", uge),
col=c("red", "red", "black", "black"), lty=c(1,2,1,2), cex=1.3)
dev.off()
女性(sex=2)のほうが男性(sex=1)よりも時間が長いことがわかります。
in_f <- "sample49.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Male", "Female")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
「GN or AN or PKD or Other」の4種類ありますので、どれがどれかはわかりませんが、4つ分描画されているのがわかります。
in_f <- "sample49.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue", "pink")
param_legend <- c("AN", "GN", "Other", "PKD")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~disease
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
男女と病気の種類の計8通り分ありますので、どれがどれかはわかりませんが、計8つ分描画されているのがわかります。
色の名前を8種類も打つのが面倒なのでデフォルトの色番号(1:black, 2:red, ...)で指定しています。
in_f <- "sample49.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- 1:8
param_legend <- c("Male_AN", "Male_GN", "Male_Other", "Male_PKD", "Female_AN", "Female_GN", "Female_Other", "Female_PKD")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex+disease
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(unknown unit)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
Timeが2500くらいまでは、890人いる女性(sex=0)のほうが968人いる男性(sex=1)よりも生存時間が短いことがわかります。
in_f <- "sample50.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("Female", "Male")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~sex
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
リンパ節転移が少ないほう(node4=0)が生存率が高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black")
param_legend <- c("node4(less)", "node4(more)")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~node4
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
経過観察のみのObs群、Levamisole単独投与群(Lev群)、そして2種類の抗がん剤の併用群(Lev+5-FU群)の3種類の比較です。
併用群の生存率が一番高いことが分かります。
in_f <- "sample50.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
param_mar <- c(4, 5, 1, 0)
param_col <- c("red", "black", "blue")
param_legend <- c("Lev", "Lev+5FU", "Obs")
param_confint <- 0.8
library(survival)
data <- read.table(in_f, header=TRUE, sep="\t")
head(data)
dim(data)
hoge <- Surv(time=time, event=status)~rx
sf <- survfit(formula=hoge, data=data, conf.int=param_confint)
sf
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(sf, xlab="Time(in days)", ylab="Survival rate",
cex.lab=1.5, cex.axis=1.5, col=param_col, conf.int=T)
grid(col="gray", lty="dotted")
legend("topright", legend=param_legend,
col=param_col, lty=1, cex=1.5)
dev.off()
2群間(A群 vs. B群)の発現変動解析結果を視覚化する手段の1つです。縦軸がlog2(B/A)またはlog2(A/B)、横軸がlog2((A+B)/2)みたいな感じで散布図をプロットします。
縦軸は、いわゆるlog ratioとかlog比などと呼ばれるものです。横軸は平均発現量という理解でよいです。数が大きいほど全体的な発現量が高い遺伝子だと判断します。
縦軸が0に近いものほど発現変動の度合いが低い(変動していない)と判断します。
ほとんどの発現変動解析用パッケージ中で、このプロットを描くための関数が用意されています。
M-A plotは、二群間比較の発現変動解析結果の図としてよく見かけます。横軸を平均発現レベル、縦軸をlog ratioとしてプロットしたものです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「G1群 5サンプル vs. G2群 5サンプル」の2群間比較データです。
in_f <- "SupplementaryTable2_changed.txt"
param_G1 <- 5
param_G2 <- 5
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
param3 <- 1
abline(h=param3, col="red", lwd=1)
サイズを指定してpng形式ファイルで保存するやり方です。
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge2.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(600, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
grid()
dev.off()
y軸の範囲をparam5で指定するやり方です。
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge3.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(600, 400)
param5 <- c(-5, 5)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1, ylim=param5)
dev.off()
グリッドを、param6で指定した色およびparam7で指定した線のタイプで表示させるやり方です。
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge4.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(600, 400)
param5 <- c(-5, 5)
param6 <- "gray"
param7 <- "solid"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1, ylim=param5)
grid(col=param6, lty=param7)
dev.off()
param8で指定した任意のIDがどのあたりにあるかをparam9で指定した色で表示させるやり方です。
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge5.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(600, 400)
param5 <- c(-8, 8)
param6 <- "gray"
param7 <- "dotted"
param8 <- c("ENSG00000004468","ENSG00000182325","ENSG00000110492","ENSG00000008516","ENSG00000100170","ENSG00000173698","ENSG00000171433")
param9 <- "red"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
obj <- is.element(rownames(data), param8)
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1, ylim=param5)
grid(col=param6, lty=param7)
points(x_axis[obj], y_axis[obj], col=param9)
dev.off()
data[obj,]
param8で指定した原著論文中でRT-PCRで発現変動が確認された7遺伝子のGene symbolsがどのあたりにあるかをparam9で指定した色で表示させるやり方です。
一見ややこしくて回りくどい感じですが、以下のような事柄に対処するために、ここで記述しているような集合演算テクニック(intersect, is.element (or %in%))を駆使することは非常に大事です:
- 一つのgene symbolが複数のEnsembl Gene IDsに対応することがよくある。
- BioMartなどから取得したIDの対応関係情報を含むアノテーションファイル(ens_gene_48.txt)中に、原著論文で言及されparam8で指定したgene symbolsが存在しない。
- 読み込んだ発現データファイル中にはあるがアノテーションファイル中には存在しないIDがある。
in_f1 <- "SupplementaryTable2_changed.txt"
in_f2 <- "ens_gene_48.txt"
out_f <- "hoge6.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(600, 400)
param5 <- c(-8, 8)
param6 <- "gray"
param7 <- "dotted"
param8 <- c("MMP25","SLC5A1","MDK","GPR64","CD38","GLOD5","FBXL6")
param9 <- "red"
data <- read.table(in_f1, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tmp <- read.table(in_f2, header=TRUE, sep="\t", quote="")
gs_annot <- tmp[,5]
names(gs_annot) <- tmp[,1]
gs_annot_sub <- intersect(gs_annot, param8)
obj_annot <- is.element(gs_annot, gs_annot_sub)
ensembl_annot_gs <- unique(names(gs_annot)[obj_annot])
ensembl_data_gs <- intersect(rownames(data), ensembl_annot_gs)
obj <- is.element(rownames(data), ensembl_data_gs)
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1, ylim=param5)
grid(col=param6, lty=param7)
points(x_axis[obj], y_axis[obj], col=param9)
dev.off()
ensembl_data_gs
data[obj,]
RPM正規化してから作図するやり方です。
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge7.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(600, 400)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
RPM <- sweep(data, 2, 1000000/colSums(data), "*")
data <- RPM
mean_G1 <- log2(apply(as.matrix(data[,data.cl==1]), 1, mean))
mean_G2 <- log2(apply(as.matrix(data[,data.cl==2]), 1, mean))
x_axis <- (mean_G1 + mean_G2)/2
y_axis <- mean_G2 - mean_G1
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(x_axis, y_axis, xlab="A = (log2(G2)+log2(G1))/2", ylab="M = log2(G2)-log2(G1)", pch=20, cex=.1)
grid()
dev.off()
「解析 | 発現変動 | 2群間 | 対応なし | 複製なし | TCC(Sun_2013)」の例題とほぼ同じですが、
TCCを用いて発現変動遺伝子(DEG)検出を行った後、
それを様々な閾値で色分けして表示させるやり方を示します。このような作業を行うことで、例題5が示すように
「何かおかしな結果になっている!バグがあるのではないか?!」といったことがわかります(南茂隆生 氏提供情報)。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
param_FDRで指定したFDR(false discovery rate)閾値を満たすDEGをマゼンタ色にしてpngファイルに保存する例です。
複製(反復)なしデータの場合は、一般的なFDR閾値(5%や10%)を満たす遺伝子は通常得られません。
このデータの場合も有意なDEGはないため、全て黒色のプロットになります。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge1.png"
param_G1 <- 1
param_G2 <- 1
param_FDR <- 0.05
param_fig <- c(400, 380)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
sum(result$q.value < param_FDR)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(FDR ", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sum(tcc$stat$q.value < 0.05)
sum(tcc$stat$q.value < 0.10)
sum(tcc$stat$q.value < 0.20)
sum(tcc$stat$q.value < 0.30)
param_pvalueで指定したFPR(false positive rate;有意水準のことです)閾値を満たすDEGを色付けしてpngファイルに保存する例です。
TCCパッケージ中のplot関数はFDR閾値でしか指定できない仕様になっているので、ここはアリエナイほど厳しい閾値(0.00000000000001)にしておいて、
後でpoints関数を用いてp-value閾値を満たすものを"skyblue"で表示させるようにしています。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge2.png"
param_G1 <- 1
param_G2 <- 1
param_pvalue <- 0.05
param_fig <- c(400, 380)
param_col <- "skyblue"
param_cex <- 1
param_pch <- 20
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=0.1)
result <- getResult(tcc, sort=FALSE)
sum(result$p.value < param_pvalue)
sum(tcc$stat$p.value < param_pvalue)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=0.00000000000001, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(p-value < ", param_pvalue, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
obj <- as.logical(tcc$stat$p.value < param_pvalue)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
sum(tcc$stat$p.value < 0.05)
sum(tcc$stat$p.value < 0.10)
sum(tcc$stat$p.value < 0.20)
sum(tcc$stat$p.value < 0.30)
param_FCで指定した変動幅以上の遺伝子を色付けしてpngファイルに保存する例です。
TCCパッケージ中のplot関数はFDR閾値でしか指定できない仕様になっているので、ここはアリエナイほど厳しい閾値(0.00000000000001)にしておいて、
後でpoints関数を用いてparam_FC倍という閾値を満たすものを"skyblue"で表示させるようにしています。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge3.png"
param_G1 <- 1
param_G2 <- 1
param_FC <- 2
param_fig <- c(400, 380)
param_col <- "skyblue"
param_cex <- 1
param_pch <- 20
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=0.1)
result <- getResult(tcc, sort=FALSE)
sum(abs(result$m.value) >= log2(param_FC))
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=0.00000000000001, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("DEG(", param_FC, "-fold)", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
obj <- as.logical(abs(result$m.value) >= log2(param_FC))
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
例題2と似ていますが、param_DEGで指定したp-valueの低い上位500個の遺伝子を"skyblue"で表示させる例です。
in_f <- "data_hypodata_1vs1.txt"
out_f <- "hoge4.png"
param_G1 <- 1
param_G2 <- 1
param_DEG <- 500
param_fig <- c(400, 380)
param_col <- "skyblue"
param_cex <- 1
param_pch <- 20
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=0.1)
result <- getResult(tcc, sort=FALSE)
ranking <- rank(tcc$stat$p.value)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=0.00000000000001, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("Top ", param_DEG, "genes", sep=""), "Others"),
col=c(param_col, "black"), pch=20)
obj <- as.logical(ranking <= param_DEG)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
例題4ととの違いは入力ファイルのみです。M-A plotが変なことがわかります。つまりDEG検出結果がオカシイということです(南茂隆生 氏提供情報)。
例えばM-A plotのM値が-5.05、A値が4.29の"FBgn0024288"は、p値が0.027で96位です。
しかし、同程度のM値(-4.97)でA値(=8.38)が非常に高い"FBgn0039155"のp値がなぜか0.395と高めに出ており2247位となっています。
内部的に用いているDESeq2の出力結果の解説(?results)ページ中でも
「outlierを判定されたもののp値はNAとしている(results assigns a p-value of NA to genes containing count outliers)」
と書いてはいますが、"FBgn0039155"のp値はNAにもなっていないのでoutlierと判定されたわけでもなさそうです。
従って、2018.01.12現在の私の見解は「DESeq2にバグがある」です。
尚、この結論に至る手前のBioconductor上でのDESeq2開発者とのやり取りはこれです。
in_f <- "sample_pasilla_1vs1.txt"
out_f <- "hoge5.png"
param_G1 <- 1
param_G2 <- 1
param_DEG <- 500
param_fig <- c(400, 380)
param_col <- "skyblue"
param_cex <- 1
param_pch <- 20
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=0.1)
result <- getResult(tcc, sort=FALSE)
ranking <- rank(tcc$stat$p.value)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=0.00000000000001, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("Top ", param_DEG, "genes", sep=""), "Others"),
col=c(param_col, "black"), pch=20)
obj <- as.logical(ranking <= param_DEG)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
sum(result$m.value < -4.5)
obj <- (result$m.value < -4.5)
result[obj, ]
例題5と基本的に同じですが、M-A plot以外の解析結果ファイルを出力させています。
in_f <- "sample_pasilla_1vs1.txt"
out_f1 <- "hoge6.txt"
out_f2 <- "hoge6.png"
param_G1 <- 1
param_G2 <- 1
param_DEG <- 500
param_fig <- c(400, 380)
param_col <- "skyblue"
param_cex <- 1
param_pch <- 20
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="deseq2", test.method="deseq2",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="deseq2", FDR=0.1)
result <- getResult(tcc, sort=FALSE)
ranking <- rank(tcc$stat$p.value)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=0.00000000000001, xlim=c(-3, 13), ylim=c(-10, 10))
legend("bottomright", c(paste("Top ", param_DEG, "genes", sep=""), "Others"),
col=c(param_col, "black"), pch=20)
obj <- as.logical(ranking <= param_DEG)
points(result$a.value[obj], result$m.value[obj],
pch=param_pch, cex=param_cex, col=param_col)
dev.off()
tmp <- cbind(rownames(tcc$count), tcc$count, result)
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
ここではサンプルデータ2のSupplementaryTable2_changed.txtの
「G1群 5サンプル vs. G2群 5サンプル」の二群間比較データ(raw count; 特定の遺伝子領域にいくつリードがマップされたかをただカウントした数値データ)
とTCCパッケージから生成したシミュレーションデータとを用いて2例を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. raw countのデータ
in_f <- "SupplementaryTable2_changed.txt"
out_f <- "hoge1.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(380, 420)
library(ggplot2)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
colSums(data)
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
hoge <- data[,data.cl==1]
nf <- mean(colSums(hoge))/colSums(hoge)
G1 <- sweep(hoge, 2, nf, "*")
colSums(G1)
hoge <- data[,data.cl==2]
nf <- mean(colSums(hoge))/colSums(hoge)
G2 <- sweep(hoge, 2, nf, "*")
colSums(G2)
mean_G1 <- log2(apply(G1, 1, mean))
mean_G2 <- log2(apply(G2, 1, mean))
df <- data.frame(
A = (mean_G1 + mean_G2) / 2,
M = mean_G2 - mean_G1
)
head(df)
l <- ggplot(df, aes(x = A, y = M))
l <- l + geom_point(size = 2, pch = 20, na.rm = T)
l <- l + xlab("A value")
l <- l + ylab("M value")
plot(l)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(l)
dev.off()
2. シミュレーションデータ
TCCパッケージで生成される2群間比較用のシミュレーションデータを描きます。
シミュレーションデータの場合G1群で高発現しているDEG (DEG1)か、G2群で高発現しているDEG (DEG2)か、
non-DEGかわかっているため、それぞれ別々の色に塗ります。2021年9月5日に、実際にはDEG1とDEG2の塗分けができていないことの指摘や修正コードをいただいたので反映させました(Manon Makino氏提供情報)。
out_f <- "hoge2.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(380, 420)
library(TCC)
library(ggplot2)
tcc <- simulateReadCounts(replicates = c(param_G1, param_G2))
ma <- plot(tcc)
tag <- tcc$simulation$trueDEG
tag[1801:2000] <- 2
tag[tag == 0] <- "non-DEG"
tag[tag == 1] <- "DEG (G1)"
tag[tag == 2] <- "DEG (G2)"
df <- data.frame(
A = ma$a.value,
M = ma$m.value,
TYPE = as.factor(tag)
)
l <- ggplot(df, aes(x = A, y = M))
l <- l + geom_point(aes(colour = TYPE), size = 2, pch = 20, na.rm = T)
l <- l + xlab("A value")
l <- l + ylab("M value")
plot(l)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(l)
dev.off()
3. シミュレーションデータ
例題2と基本的に同じですが、プロットしたときにnon-DEGが後に描画される(これが例題2)のではなく、
DEGが後に描画されるようにデータを並べ替える操作を行った版です(Manon Makino氏提供情報)。
out_f <- "hoge3.png"
param_G1 <- 5
param_G2 <- 5
param_fig <- c(380, 420)
library(TCC)
library(ggplot2)
tcc <- simulateReadCounts(replicates = c(param_G1, param_G2))
ma <- plot(tcc)
tag <- tcc$simulation$trueDEG
tag[1801:2000] <- 2
tag[tag == 0] <- "non-DEG"
tag[tag == 1] <- "DEG (G1)"
tag[tag == 2] <- "DEG (G2)"
df <- data.frame(
A = ma$a.value,
M = ma$m.value,
TYPE = as.factor(tag)
)
df <- rbind(df[df$TYPE=='non-DEG',],
df[df$TYPE=='DEG (G1)',],
df[df$TYPE=='DEG (G2)',])
l <- ggplot(df, aes(x = A, y = M))
l <- l + geom_point(aes(colour = TYPE), size = 2, pch = 20, na.rm = T)
l <- l + xlab("A value")
l <- l + ylab("M value")
plot(l)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(l)
dev.off()
遺伝子発現行列のような数値行列を入力として、類似した発現パターンを示す列(サンプル)をまとめた結果を描画するやり方ですが、
基本的に、「解析 | クラスタリング | ...」のほうをご覧ください。
TCCパッケージ中の
clusterSample関数を利用したサンプル間クラスタリング結果をいろいろ整形するやり方を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
Neyret-Kahn et al., Genome Res., 2013の2群間比較用ヒトRNA-seqカウントデータです。
パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)から得られます。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge1.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
2. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
1.との違いはplot関数実行時のオプションの部分のみです。下のほうの「hclust(*, "average")」という文字が消えていることが分かります。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge2.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, sub="")
dev.off()
3. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
2.との違いはplot関数実行時のオプションの部分のみです。下のほうの「d」という文字が消えていることが分かります。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge3.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, sub="", xlab="")
dev.off()
4. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
3.との違いはplot関数実行時のオプションの部分のみです。表示されているy軸名「Height」の文字の大きさを通常の1.5倍にしています。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge4.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, sub="", xlab="", cex.lab=1.5)
dev.off()
5. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
4.との違いはplot関数実行時のオプションの部分のみです。
表示されているy軸名「Height」の文字の大きさを通常の0.6倍にし、入力ファイル中のサンプル名の文字の大きさを通常の1.3倍にしています。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge5.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, sub="", xlab="", cex.lab=0.6, cex=1.3)
dev.off()
6. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
5.との違いはplot関数実行時のオプションの部分のみです。
y軸名を通常の1.2倍の「height kamo」に、そして図の上のほうのタイトル名が表示されないようにしてます。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge6.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="height kamo")
dev.off()
7. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
6.との違いはpar関数部分のみです。図の上下の余白を狭くして、図の左側のみ4行分空け、それ以外を1行分だけ開けるように指定しています。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge7.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(1, 4, 1, 1))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="height kamo")
dev.off()
8. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
7.との違いはpar関数実行時のオプションの部分のみです。
図の上下の余白を狭くして、図の左側のみ4行分、上を1行分空け、下と右を0行分だけ開けるように指定しています。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge8.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="height kamo")
dev.off()
9. 59,857 genes×6 samplesのリアルデータ(srp017142_count_bowtie.txt; 3 proliferative samples vs. 3 Ras samples)の場合:
8.との違いはpar関数実行時のオプションの部分のみです。
縦軸(y軸)の範囲はデフォルトでよきに計らってくれるので、8の結果から0.06未満であることがわかりますが、
ylim=c(0, 0.2)で縦軸の範囲を0から0.2と明示的に指定していますがうまく反映されないようです(爆)。
in_f <- "srp017142_count_bowtie.txt"
out_f <- "hoge9.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(0, 4, 1, 0))
plot(out, sub="", xlab="", cex.lab=1.2,
cex=1.3, main="", ylab="height kamo",
ylim=c(0.0, 0.2))
dev.off()
ROC曲線(Receiver Operating Characteristic Curve)を描くパッケージです。バイオインフォマティクス分野では、
特にシミュレーションデータの性能評価指標の1つとして、ROC曲線の下部面積(Area Under the ROC curve)が使われます。
発現変動解析の手法比較系(バイオインフォマティクス系)の論文では、「真の発現変動遺伝子(true DEGs)」を予め決めておき、
どのDEG検出法が「真の発現変動遺伝子」をより上位にすることができるかを示す指標として、
「ROC曲線の下部面積(Area Under the ROC curve; AUC)」で方法の比較を行っています。このAUC値を計算するための基礎情報がROC曲線です。
必要な情報は2つのベクトルです。1つは何らかのランキング法を用いて発現変動の度合いで遺伝子をランキングした順位情報、
そしてもう1つはどの遺伝子が「真の発現変動遺伝子」かを示す0 or 1の情報です。
この項目では、全部で10遺伝子(gene1, gene2, ..., gene10)、そのうちgene1, 3, 5が真のDEGsという仮定で、様々なランキング結果でのROC曲線とAUC値、
および範囲を限定したAUC値(partial AUC)を考えます。
ここでは、ROC曲線の「横軸を"1-specificity"、縦軸を"sensitivity"」としていますが、同じ意味なので「横軸を"False Positive Rate (FPR)"、縦軸を"True Positive Rate (TPR)"」としてもよいです。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 真のDEGsに相当するgene1が2位、gene3が3位、gene5が1位の場合:
DEGsとnon-DEGsを完璧に区別できている理想的なランキング結果(AUC = 1)の例です。
param_rankで指定する際に1, 3, 5番目の要素のところのいずれかが1, 2, 3となっていればいいです。
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(2, 8, 3, 7, 1, 10, 5, 4, 9, 6)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
2. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
現実によく見かけるROC曲線やAUC値(=0.857)の一例です。
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
3. 真のDEGsに相当するgene1が2位、gene3が4位、gene5が3位の場合:
2.と同じAUC値(=0.857)ですが、ROC曲線が異なります。
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(2, 8, 4, 7, 3, 10, 5, 1, 9, 6)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
4. 真のDEGsに相当するgene1が2位、gene3が6位、gene5が1位の場合:
2.と同じAUC値(=0.857)ですが、ROC曲線が異なります。
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(2, 8, 6, 7, 1, 10, 5, 4, 9, 3)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
5. 真のDEGsに相当するgene1が2位、gene3が6位、gene5が1位の場合:
4.と同じですが、x軸が全範囲のfull AUC値(=0.857)だけでなく、param_thresで指定した範囲までに限定したpartial AUC値(=0.13333)も計算しています。
out_f <- "hoge5.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(2, 8, 6, 7, 1, 10, 5, 4, 9, 3)
param_fig <- c(400, 400)
param_thres <- 0.2
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
pAUC(out, t0 = param_thres)
6. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が10位の場合:
3つのDEGの平均順位が(1+5+10)/3 = 5.33位、7つのnon-DEGの平均順位が(2+3+4+6+7+8+9)/7 = 5.57位であることからも、full AUC値(=0.5238)が0.5付近にあることの妥当性が分かります。
out_f <- "hoge6.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 10, 2, 6, 4, 9, 3)
param_fig <- c(400, 400)
param_thres <- 0.2
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
pAUC(out, t0 = param_thres)
7. 真のDEGsに相当するgene1が8位、gene3が9位、gene5が10位の場合:
DEG検出を行っているつもりで、全non-DEGsが上位を占めている最悪のランキング結果(AUC = 0)の例です。
通常はこのようなことは起こりません。解析手順が間違っていると考えるのが妥当です。
out_f <- "hoge7.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(8, 1, 9, 7, 10, 2, 6, 4, 5, 3)
param_fig <- c(400, 400)
param_thres <- 0.2
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
pAUC(out, t0 = param_thres)
8. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
2.と基本的に同じで、ROC曲線の図を描くための感度(sensitivity)と特異度(specificity)の元の数値情報をテキストファイル形式で取得する例です。
out_f1 <- "hoge8.txt"
out_f2 <- "hoge8.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
tmp <- cbind(1 - out@spec, out@sens)
tmp <- tmp[order(tmp[, 1]), ]
colnames(tmp) <- c("1-sensitivity", "specificity")
write.table(tmp, out_f1, sep="\t", append=F, quote=F, row.names=F)
png(out_f2, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity", main="ROC curve")
grid(col="gray", lty="dotted")
dev.off()
AUC(out)
「基礎1」では、主にDEGの順位情報を変えて、ROC曲線やAUC値がどのように変わっていくのかを中心に述べました。
この項目では、プロットの色、軸の数値の色、タイトル、x軸やy軸ラベルの色を様々な手段で指定するやり方を示します。
全部で10遺伝子(gene1, gene2, ..., gene10)、そのうちgene1, 3, 5が真のDEGsという仮定はそのままです。
R Console画面上で「?par」と打つとplot関数実行時に利用可能なオプションを調べることができます。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
ROC曲線の色を赤色("red")で指定しています。デフォルトは"black"で、"green",
"blue", "orange", "yellow", "gray"などが指定できます。
R Console画面上で「colors()」と打つと、指定可能な色名をリストアップできます。
実際の色も同時に調べたい場合は「demo("colors")」と打ち込めばよい。
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity",
main="ROC curve", col=param_col)
grid(col="gray", lty="dotted")
dev.off()
2. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
色を数値で表しています。黒("black", 1), 赤("red", 2), 緑("green3", 3), 青("blue", 4), シアン("cyan", 5), マゼンタ("magenta", 6), 黄色("yellow", 7), 灰色("gray", 8)です。
R Console画面上で「palette()」と打つと、色名と要素番号の対応関係がわかりますが、8までで終わりなようです。
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- 7
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity", ylab="sensitivity",
main="ROC curve", col=param_col)
grid(col="gray", lty="dotted")
dev.off()
3. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
色をRGB (red, green, blue)の0-255の数値で指定するやり方です。
黒(0, 0, 0), 赤(255, 0, 0), 緑(0, 255, 0), 青(0, 0, 255), 黄色(255, 255, 0), マゼンタ(255, 0, 255), シアン(0, 255, 255), 白(255, 255, 255)です。
例えば、灰色でも(20, 0, 0)とすれば白に近い色となり、(200, 0, 0)とすれば黒に近い色になります。
黄緑色は黄色と緑の中間なので(123, 255, 0)、オレンジ色は赤と黄色の中間なので(255, 123, 0)のような感じで表現できます。
RGBで指定するときは、param_colの箇所以外にも、plot関数中のcolオプションのところの記述形式も変える必要がありますのでご注意ください。
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- c(255, 123, 0)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, xlab="1-specificity",
ylab="sensitivity",
main="ROC curve",
col=rgb(param_col[1], param_col[2], param_col[3], max=255))
grid(col="gray", lty="dotted")
dev.off()
4. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
ROC曲線の色をオレンジ色("orange")で指定しています。
それ以外にも、軸(col.axisオプション)、x軸とy軸のラベル(col.lab)、メインタイトル(col.main)、
ここでは出ていませんが図の下のほうにあるサブタイトル(col.sub)の色も任意に与えることができます。
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "orange"
param_col_axis <- "blue"
param_col_lab <- "cyan"
param_col_main <- "magenta"
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out,
main="ROC curve", col.main=param_col_main,
xlab="1-specificity", ylab="sensitivity",
col.lab=param_col_lab,
col.axis=param_col_axis,
col=param_col)
grid(col="gray", lty="dotted")
dev.off()
この項目では、プロット(cex)、軸の数値(cex.axis)、メインタイトル(cex.main)、x軸やy軸ラベル(cex.lab)、
サブタイトル(cex.sub)についての大きさの倍率を指定するやり方を示します。
基本的には1.0が通常の大きさで、0.5だと通常の0.5倍、
1.7だと通常の1.7倍の大きさで描画してくれます。
文字関連の大きさ調整を行うcexの他に、線分の幅を指定するlwd、線分の種類(実線やダッシュなど)を指定するltyの指定も行います。
全部で10遺伝子(gene1, gene2, ..., gene10)、そのうちgene1, 3, 5が真のDEGsという仮定はそのままです。
R Console画面上で「?par」と打つとplot関数実行時に利用可能なオプションを調べることができます。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
param_colでROC曲線の色をオレンジ色("orange")で指定している他、
軸(cex.axisオプション)、x軸とy軸のラベル(cex.lab)、メインタイトル(cex.main)、
ここでは出ていませんが図の下のほうにあるサブタイトル(cex.sub)の大きさも任意に与えることができます。
尚、以下のparam_cexは実際には機能していません。理由はここではROC曲線の線のみであり、
param_cexはプロット中のtextやsymbolの大きさを指定するものだからです。
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "orange"
param_cex <- 2.0
param_cex_axis <- 1.2
param_cex_lab <- 0.7
param_cex_main <- 2.3
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out,
main="ROC curve", cex.main=param_cex_main,
xlab="1-specificity", ylab="sensitivity",
cex.lab=param_cex_lab,
cex.axis=param_cex_axis,
cex=param_cex, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
2. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
ROC曲線の線分の幅を指定するlwdオプションの数値を変更しています。おそらくここで指定している数値は倍率に相当するものです。
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "orange"
param_lwd <- 2.5
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve",
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col)
grid(col="gray", lty="dotted")
dev.off()
3. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
ROC曲線の線分の種類をltyオプションで指定しています。colオプション同様、数値でも文字でも指定可能です。
実線("solid", 1), ダッシュ("dashed", 2), ドット("dotted", 3)あたりが基本だと思います。
他にも、透明("blank", 0), ドットとダッシュ両方("dotdash", 4), ("longdash", 5), ("twodash", 6)などがあります。
以下の例はロングダッシュを数値で指定しています。
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "orange"
param_lwd <- 4.6
param_lty <- 5
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve",
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
4. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
ROC曲線の線分の種類をltyオプションで指定しています。colオプション同様、数値でも文字でも指定可能です。
実線("solid", 1), ダッシュ("dashed", 2), ドット("dotted", 3)あたりが基本だと思います。
他にも、透明("blank", 0), ドットとダッシュ両方("dotdash", 4), ("longdash", 5), ("twodash", 6)などがあります。
以下の例は色を"blue"、幅を2.0、種類を"dashed"にしています。
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "blue"
param_lwd <- 2.0
param_lty <- "dashed"
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve",
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
この項目では、図の重ね書き時に必要なテクニックとして、annとaxesオプションの挙動を示します。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
param_colでROC曲線の色を赤色("red")、線分の幅を1.5倍、
線分の種類を実線("solid")で示す他に、main, xlab, and ylabオプションを明記しなかった場合の結果を示しています。
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out,
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
2. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
plot関数実行時に、ann=Fオプションをつけるとxlabやylabを指定していても無視されることがわかります。
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, ann=F,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
3. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
メインタイトルのみ表示させたい場合のやり方です。
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve",
xlab="", ylab="",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
4. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
axes=Fとすると軸ラベルの数値が消えます。
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve", axes=F,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
この項目では、軸ラベルの表示角度を指定するlasオプションを示します。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
las=0はデフォルト(ラベルを各軸に対して平行に描く)なので特に指定しないときと比べて変化はありません。
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
param_las <- 0
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve", las=param_las,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
2. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
las=1とすると、ラベルを全て水平に描くことができます。この場合、y軸の数値が時計回りに90度回っていることが分かります。
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
param_las <- 1
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve", las=param_las,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
3. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
las=2とすると、ラベルを各軸に対して垂直に描くことができます。特にx軸上のラベルを垂直に描けるので全て表示させたい場合に重宝します。
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
param_las <- 2
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve", las=param_las,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
4. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
las=3とすると、ラベルを全て垂直に描くことができます。
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
param_las <- 3
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out, main="ROC curve", las=param_las,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
この項目では、図の余白を指定するpar関数内で用いるmarオプションを示します。
「下、左、上、右」の順で余白を指定します。単位は「行」で、0が最も余白が小さく、大体5程度が最大です。
私の通常の使い方は「左以外は0に近い数値にする」です。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
左側以外の余白を0行にした例です。上側のメインタイトル(ROC curveの文字)が切れていることが分かります。
また、下側の軸ラベルや軸タイトルも余白の関係上消えていることがわかります。
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
param_las <- 0
param_mar <- c(0, 4, 0, 0)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(out, main="ROC curve", las=param_las,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
2. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
左側の余白を4行、下側の余白を2行にした例です。main=""として上側のメインタイトルの文字も消しています。
下側の余白が2行では、x軸タイトルの表示スペースがないことがわかります。
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
param_las <- 0
param_mar <- c(2, 4, 0, 0)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(out, main="", las=param_las,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
3. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
左側の余白を4行、下側の余白を4行、それ以外の余白を0.5行にした例です。小数点の数値も指定できるようですね。
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
param_col <- "red"
param_lwd <- 1.5
param_lty <- "solid"
param_las <- 0
param_mar <- c(4, 4, 0.5, 0.5)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=param_mar)
plot(out, main="", las=param_las,
xlab="1-specificity", ylab="sensitivity",
lwd=param_lwd, col=param_col, lty=param_lty)
grid(col="gray", lty="dotted")
dev.off()
4. 真のDEGsに相当するgene1が5位、gene3が1位、gene5が3位の場合:
3.と同じです、私の普段使いでは、図のパラメータはこのようにそのまま書いちゃいます。
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_rank <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank))
obj[param_DEG] <- 1
out <- rocdemo.sca(truth = obj, data = -param_rank)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0.5, 0.5))
plot(out, main="", las=0,
xlab="1-specificity", ylab="sensitivity",
lwd=1.5, col="red", lty="solid")
grid(col="gray", lty="dotted")
dev.off()
この項目では、図の重ね書き時に指定するpar関数内で用いるnewオプションを示します。
基本的には、par(new=T)を書くだけです。
ここでは、ランキング結果1の数値ベクトルがc(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)、
ランキング結果2がc(3, 1, 6, 2, 5, 10, 7, 4, 9, 8)、
ランキング結果3がc(4, 8, 1, 3, 2, 10, 7, 5, 9, 6)
として2つのランキング結果の重ね合わせや3つの結果の重ね合わせの例を示します。
「ファイル」−「ディレクトリの変更」でファイルを保存したいディレクトリに移動し以下をコピペ。
1. 2つのランキング結果の重ね書きを行う場合:
ランキング結果1("red")とランキング結果2("blue")の重ね合わせの基本形です。
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_rank1 <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_rank2 <- c(3, 1, 6, 2, 5, 10, 7, 4, 9, 8)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -param_rank1)
out2 <- rocdemo.sca(truth = obj, data = -param_rank2)
AUC(out1)
AUC(out2)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out1, axes=F, ann=F, col="red")
par(new=T)
plot(out2, col="blue")
dev.off()
2. 2つのランキング結果の重ね書きを行う場合:
x軸、y軸ラベル情報とグリッドを追加しています。
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_rank1 <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_rank2 <- c(3, 1, 6, 2, 5, 10, 7, 4, 9, 8)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -param_rank1)
out2 <- rocdemo.sca(truth = obj, data = -param_rank2)
AUC(out1)
AUC(out2)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out1, axes=F, ann=F, col="red")
par(new=T)
plot(out2, col="blue",
xlab="1-specificity", ylab="sensitivity")
grid(col="gray", lty="dotted")
dev.off()
3. 2つのランキング結果の重ね書きを行う場合:
図の余白を指定しているほか、lwdで線分の幅をランキング結果1(3.9倍)とランキング結果2(2.3倍)でそれぞれ指定しています。
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_rank1 <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_rank2 <- c(3, 1, 6, 2, 5, 10, 7, 4, 9, 8)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -param_rank1)
out2 <- rocdemo.sca(truth = obj, data = -param_rank2)
AUC(out1)
AUC(out2)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0.3, 0.3))
plot(out1, axes=F, ann=F,
col="red", lwd=3.9)
par(new=T)
plot(out2, col="blue", lwd=2.3,
xlab="1-specificity", ylab="sensitivity")
grid(col="gray", lty="dotted")
dev.off()
4. 3つのランキング結果の重ね書きを行う場合:
ランキング結果1("red")とランキング結果2("blue")とランキング結果3("magenta")の重ね合わせの基本形です。
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_rank1 <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_rank2 <- c(3, 1, 6, 2, 5, 10, 7, 4, 9, 8)
param_rank3 <- c(4, 8, 1, 3, 2, 10, 7, 5, 9, 6)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -param_rank1)
out2 <- rocdemo.sca(truth = obj, data = -param_rank2)
out3 <- rocdemo.sca(truth = obj, data = -param_rank3)
AUC(out1)
AUC(out2)
AUC(out3)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out1, axes=F, ann=F, col="red")
par(new=T)
plot(out2, axes=F, ann=F, col="blue")
par(new=T)
plot(out3, col="magenta")
dev.off()
5. 3つのランキング結果の重ね書きを行う場合:
図の余白、ltyで線分の種類、lwdで線分の幅などいろいろ指定しています。
out_f <- "hoge5.png"
param_DEG <- c(1, 3, 5)
param_rank1 <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_rank2 <- c(3, 1, 6, 2, 5, 10, 7, 4, 9, 8)
param_rank3 <- c(4, 8, 1, 3, 2, 10, 7, 5, 9, 6)
param_fig <- c(400, 400)
library(ROC)
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -param_rank1)
out2 <- rocdemo.sca(truth = obj, data = -param_rank2)
out3 <- rocdemo.sca(truth = obj, data = -param_rank3)
AUC(out1)
AUC(out2)
AUC(out3)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(out1, axes=F, ann=F,
col="red", lwd=2.0, lty="dotted")
par(new=T)
plot(out2, axes=F, ann=F,
col="blue", lwd=3.0, lty="dashed")
par(new=T)
plot(out3, col="magenta",
lwd=1.0, lty="solid",
xlab="1-specificity", ylab="sensitivity")
grid(col="gray", lty="dotted")
dev.off()
6. 3つのランキング結果の重ね書きを行う場合:
5.と基本的に同じです。このくらいの数になってくるとparamのところで最初に書き込んでおくほうがミスが少なくなります。
cex.lab=1.5は軸ラベルの文字の大きさを通常の1.5倍にするという意味です。
out_f <- "hoge6.png"
param_DEG <- c(1, 3, 5)
param_rank1 <- c(5, 8, 1, 7, 3, 10, 2, 4, 9, 6)
param_rank2 <- c(3, 1, 6, 2, 5, 10, 7, 4, 9, 8)
param_rank3 <- c(4, 8, 1, 3, 2, 10, 7, 5, 9, 6)
param_fig <- c(400, 400)
param_col <- c("red", "blue", "magenta")
param_lwd <- c(2.0, 3.0, 1.0)
param_lty <- c("dotted", "dashed", "solid")
library(ROC)
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -param_rank1)
out2 <- rocdemo.sca(truth = obj, data = -param_rank2)
out3 <- rocdemo.sca(truth = obj, data = -param_rank3)
AUC(out1)
AUC(out2)
AUC(out3)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(out1, axes=F, ann=F,
col=param_col[1], lwd=param_lwd[1], lty=param_lty[1])
par(new=T)
plot(out2, axes=F, ann=F,
col=param_col[2], lwd=param_lwd[2], lty=param_lty[2])
par(new=T)
plot(out3, col=param_col[3],
lwd=param_lwd[3], lty=param_lty[3],
xlab="1-specificity", ylab="sensitivity",
cex.lab=1.5)
grid(col="gray", lty="dotted")
dev.off()
7. 3つのランキング結果の重ね書きを行う場合:
6.と基本的に同じです。ランキング結果のタブ区切りテキストファイル(roc_ranking.txt)から読み込むやり方です。
in_f <- "roc_ranking.txt"
out_f <- "hoge7.png"
param_DEG <- c(1, 3, 5)
param_fig <- c(400, 400)
param_col <- c("red", "blue", "magenta")
param_lwd <- c(2.0, 3.0, 1.0)
param_lty <- c("dotted", "dashed", "solid")
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -data[, 1])
out2 <- rocdemo.sca(truth = obj, data = -data[, 2])
out3 <- rocdemo.sca(truth = obj, data = -data[, 3])
AUC(out1)
AUC(out2)
AUC(out3)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(out1, axes=F, ann=F,
col=param_col[1], lwd=param_lwd[1], lty=param_lty[1])
par(new=T)
plot(out2, axes=F, ann=F,
col=param_col[2], lwd=param_lwd[2], lty=param_lty[2])
par(new=T)
plot(out3, col=param_col[3],
lwd=param_lwd[3], lty=param_lty[3],
xlab="1-specificity", ylab="sensitivity",
cex.lab=1.5)
grid(col="gray", lty="dotted")
dev.off()
この項目では、凡例を追加するlegend関数の基本的な使用法を示します。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
1. 3つのランキング結果のタブ区切りテキストファイル(roc_ranking.txt)の場合:
入力ファイル中にランキング法の名前が書きこまれているのでそれを利用しています。
ROC曲線は右上には必ず曲線が描かれるので、右下("bottomright")に描いています。
bottomright以外に"bottom", "bottomleft", "left", "topleft", "top", "topright", "right", "center"を指定可能です。
in_f <- "roc_ranking.txt"
out_f <- "hoge1.png"
param_DEG <- c(1, 3, 5)
param_fig <- c(400, 400)
param_col <- c("red", "blue", "magenta")
param_lwd <- c(2.0, 3.0, 1.0)
param_lty <- c("dotted", "dashed", "solid")
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -data[, 1])
out2 <- rocdemo.sca(truth = obj, data = -data[, 2])
out3 <- rocdemo.sca(truth = obj, data = -data[, 3])
AUC(out1)
AUC(out2)
AUC(out3)
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(out1, axes=F, ann=F,
col=param_col[1], lwd=param_lwd[1], lty=param_lty[1])
par(new=T)
plot(out2, axes=F, ann=F,
col=param_col[2], lwd=param_lwd[2], lty=param_lty[2])
par(new=T)
plot(out3, col=param_col[3],
lwd=param_lwd[3], lty=param_lty[3],
xlab="1-specificity", ylab="sensitivity",
cex.lab=1.5)
grid(col="gray", lty="dotted")
legend(x="bottomright",
legend=colnames(data),
col=param_col,
lwd=param_lwd,
lty=param_lty,
merge=T)
dev.off()
2. 3つのランキング結果のタブ区切りテキストファイル(roc_ranking.txt)の場合:
legendの位置を右側("right")にし、AUC値も一緒に凡例の中に示すやり方の基本形です。
AUC値の小数点以下の桁数がそのまま表示されています。また、方法名とAUC値の間に"_"を入れています。
in_f <- "roc_ranking.txt"
out_f <- "hoge2.png"
param_DEG <- c(1, 3, 5)
param_fig <- c(400, 400)
param_col <- c("red", "blue", "magenta")
param_lwd <- c(2.0, 3.0, 1.0)
param_lty <- c("dotted", "dashed", "solid")
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -data[, 1])
out2 <- rocdemo.sca(truth = obj, data = -data[, 2])
out3 <- rocdemo.sca(truth = obj, data = -data[, 3])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(out1, axes=F, ann=F,
col=param_col[1], lwd=param_lwd[1], lty=param_lty[1])
par(new=T)
plot(out2, axes=F, ann=F,
col=param_col[2], lwd=param_lwd[2], lty=param_lty[2])
par(new=T)
plot(out3, col=param_col[3],
lwd=param_lwd[3], lty=param_lty[3],
xlab="1-specificity", ylab="sensitivity",
cex.lab=1.5)
grid(col="gray", lty="dotted")
auc <- c(AUC(out1), AUC(out2), AUC(out3))
colnames(data)
hoge <- paste(colnames(data), auc, sep="_")
legend(x="right",
legend=hoge,
col=param_col,
lwd=param_lwd,
lty=param_lty,
merge=T)
dev.off()
3. 3つのランキング結果のタブ区切りテキストファイル(roc_ranking.txt)の場合:
legendの位置を右側("bottomright")にし、「方法名(AUC=XX)」のような書き方にする基本形です。
in_f <- "roc_ranking.txt"
out_f <- "hoge3.png"
param_DEG <- c(1, 3, 5)
param_fig <- c(400, 400)
param_col <- c("red", "blue", "magenta")
param_lwd <- c(2.0, 3.0, 1.0)
param_lty <- c("dotted", "dashed", "solid")
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -data[, 1])
out2 <- rocdemo.sca(truth = obj, data = -data[, 2])
out3 <- rocdemo.sca(truth = obj, data = -data[, 3])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(out1, axes=F, ann=F,
col=param_col[1], lwd=param_lwd[1], lty=param_lty[1])
par(new=T)
plot(out2, axes=F, ann=F,
col=param_col[2], lwd=param_lwd[2], lty=param_lty[2])
par(new=T)
plot(out3, col=param_col[3],
lwd=param_lwd[3], lty=param_lty[3],
xlab="1-specificity", ylab="sensitivity",
cex.lab=1.5)
grid(col="gray", lty="dotted")
auc <- c(AUC(out1), AUC(out2), AUC(out3))
hoge <- paste(colnames(data), "(AUC=", auc, ")", sep="")
hoge
legend(x="bottomright",
legend=hoge,
col=param_col,
lwd=param_lwd,
lty=param_lty,
merge=T)
dev.off()
4. 3つのランキング結果のタブ区切りテキストファイル(roc_ranking.txt)の場合:
AUC値の表示桁数を小数点以下3桁までにするやり方です。
in_f <- "roc_ranking.txt"
out_f <- "hoge4.png"
param_DEG <- c(1, 3, 5)
param_fig <- c(400, 400)
param_col <- c("red", "blue", "magenta")
param_lwd <- c(2.0, 3.0, 1.0)
param_lty <- c("dotted", "dashed", "solid")
library(ROC)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
obj <- rep(0, length(param_rank1))
obj[param_DEG] <- 1
out1 <- rocdemo.sca(truth = obj, data = -data[, 1])
out2 <- rocdemo.sca(truth = obj, data = -data[, 2])
out3 <- rocdemo.sca(truth = obj, data = -data[, 3])
png(out_f, pointsize=13, width=param_fig[1], height=param_fig[2])
par(mar=c(4, 4, 0, 0))
plot(out1, axes=F, ann=F,
col=param_col[1], lwd=param_lwd[1], lty=param_lty[1])
par(new=T)
plot(out2, axes=F, ann=F,
col=param_col[2], lwd=param_lwd[2], lty=param_lty[2])
par(new=T)
plot(out3, col=param_col[3],
lwd=param_lwd[3], lty=param_lty[3],
xlab="1-specificity", ylab="sensitivity",
cex.lab=1.5)
grid(col="gray", lty="dotted")
auc <- c(AUC(out1), AUC(out2), AUC(out3))
sprintf("%.4f", auc)
sprintf("%6.2f", auc)
hoge <- paste(colnames(data), "(AUC=", sprintf("%.3f", auc), ")", sep="")
hoge
legend(x="bottomright",
legend=hoge,
col=param_col,
lwd=param_lwd,
lty=param_lty,
merge=T)
dev.off()
私の2008,2009年の論文では、
主にRT-PCRで発現変動が確認された遺伝子を「真の発現変動遺伝子(true DEGs)」とし、どの発現変動遺伝子のランキング法が「真の発現変動遺伝子」をより上位にすることができるかを示す指標として、
「ROC曲線の下部面積(Area Under the ROC curve; AUC)」で方法の比較を行っています。このAUC値を計算するための基礎情報がROC曲線です。
必要な情報は2つのベクトルです。1つは何らかのランキング法を用いて発現変動の度合いで遺伝子をランキングした順位情報、
そしてもう1つはどの遺伝子が「真の発現変動遺伝子」かを示す0 or 1の情報です。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
「G1群 5サンプル vs. G2群 5サンプル」の2群間比較データです。「真の発現変動遺伝子」の情報をparam3で指定するやり方です。
in_f <- "SupplementaryTable2_changed.txt"
param_G1 <- 5
param_G2 <- 5
param3 <- c("ENSG00000004468","ENSG00000182325","ENSG00000110492","ENSG00000008516","ENSG00000100170","ENSG00000173698","ENSG00000171433")
param4 <- "False Positive Rate (FPR)"
param5 <- "True Positive Rate (TPR)"
param6 <- "ROC curves for raw count data"
library(ROC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
groups <- list(NDE=rep(1, (param_G1+param_G2)), DE=data.cl)
data1 <- new("countData", data=as.matrix(data), replicates=data.cl, libsizes=as.integer(colSums(data)), groups=groups)
data1P.NB <- getPriors.NB(data1, samplesize=1000, estimation="QL", cl=NULL)
out <- getLikelihoods.NB(data1P.NB, pET="BIC", cl=NULL)
out@estProps
stat_bayseq <- out@posteriors[,2]
rank_bayseq <- rank(-stat_bayseq, ties.method="min")
obj <- is.element(rownames(data), param3)
obj[obj == "TRUE"] <- 1
out <- rocdemo.sca(truth = obj, data =-rank_bayseq)
plot(out, xlab=param4, ylab=param5, main=param6)
AUC(out)
2. 「1.を基本としつつ、さらにもう一つのランキング法を実行して、二つのROC曲線を重ね書きしたい」場合:
in_f <- "SupplementaryTable2_changed.txt"
param_G1 <- 5
param_G2 <- 5
param3 <- c("ENSG00000004468","ENSG00000182325","ENSG00000110492","ENSG00000008516","ENSG00000100170","ENSG00000173698","ENSG00000171433")
param4 <- "False Positive Rate (FPR)"
param5 <- "True Positive Rate (TPR)"
param6 <- "ROC curves for raw count data"
source("http://www.iu.a.u-tokyo.ac.jp/~kadota/R/R_functions.R")
library(ROC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
groups <- list(NDE=rep(1, (param_G1+param_G2)), DE=data.cl)
data1 <- new("countData", data=as.matrix(data), replicates=data.cl, libsizes=as.integer(colSums(data)), groups=groups)
data1P.NB <- getPriors.NB(data1, samplesize=1000, estimation="QL", cl=NULL)
out <- getLikelihoods.NB(data1P.NB, pET="BIC", cl=NULL)
out@estProps
stat_bayseq <- out@posteriors[,2]
rank_bayseq <- rank(-stat_bayseq, ties.method="min")
datalog <- log2(data + 1)
stat_AD <- AD(data=datalog, data.cl=data.cl)
rank_AD <- rank(-abs(stat_AD), ties.method="min")
obj <- is.element(rownames(data), param3)
obj[obj == "TRUE"] <- 1
out1 <- rocdemo.sca(truth = obj, data =-rank_bayseq)
plot(out1, axes=F, ann=F)
par(new=T)
out2 <- rocdemo.sca(truth = obj, data =-rank_AD)
plot(out2, xlab=param4, ylab=param5, main=param6)
AUC(out)
3. 「2.を基本としつつ、ランキング法ごとに指定した色にしたい」場合:
in_f <- "SupplementaryTable2_changed.txt"
param_G1 <- 5
param_G2 <- 5
param3 <- c("ENSG00000004468","ENSG00000182325","ENSG00000110492","ENSG00000008516","ENSG00000100170","ENSG00000173698","ENSG00000171433")
param4 <- "False Positive Rate (FPR)"
param5 <- "True Positive Rate (TPR)"
param6 <- "ROC curves for raw count data"
param7 <- c( 0, 0, 0)
param8 <- c(255, 0, 0)
source("http://www.iu.a.u-tokyo.ac.jp/~kadota/R/R_functions.R")
library(ROC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
groups <- list(NDE=rep(1, (param_G1+param_G2)), DE=data.cl)
data1 <- new("countData", data=as.matrix(data), replicates=data.cl, libsizes=as.integer(colSums(data)), groups=groups)
data1P.NB <- getPriors.NB(data1, samplesize=1000, estimation="QL", cl=NULL)
out <- getLikelihoods.NB(data1P.NB, pET="BIC", cl=NULL)
out@estProps
stat_bayseq <- out@posteriors[,2]
rank_bayseq <- rank(-stat_bayseq, ties.method="min")
datalog <- log2(data + 1)
stat_AD <- AD(data=datalog, data.cl=data.cl)
rank_AD <- rank(-abs(stat_AD), ties.method="min")
obj <- is.element(rownames(data), param3)
obj[obj == "TRUE"] <- 1
out1 <- rocdemo.sca(truth = obj, data =-rank_bayseq)
plot(out1, axes=F, ann=F, col=rgb(param7[1], param7[2], param7[3], max=255))
par(new=T)
out2 <- rocdemo.sca(truth = obj, data =-rank_AD)
plot(out2, xlab=param4, ylab=param5, main=param6, col=rgb(param8[1], param8[2], param8[3], max=255))
AUC(out1)
AUC(out2)
4. 「3.を基本としつつ、legendも追加したい(ここではとりあえず「lwd=1」としてますが線分の形式をいろいろ変えることができます(詳細はこちら))」場合:
in_f <- "SupplementaryTable2_changed.txt"
param_G1 <- 5
param_G2 <- 5
param3 <- c("ENSG00000004468","ENSG00000182325","ENSG00000110492","ENSG00000008516","ENSG00000100170","ENSG00000173698","ENSG00000171433")
param4 <- "False Positive Rate (FPR)"
param5 <- "True Positive Rate (TPR)"
param6 <- "ROC curves for raw count data"
param7 <- c( 0, 0, 0)
param8 <- c(255, 0, 0)
param9 <- "baySeq"
param10 <- "AD"
source("http://www.iu.a.u-tokyo.ac.jp/~kadota/R/R_functions.R")
library(ROC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
groups <- list(NDE=rep(1, (param_G1+param_G2)), DE=data.cl)
data1 <- new("countData", data=as.matrix(data), replicates=data.cl, libsizes=as.integer(colSums(data)), groups=groups)
data1P.NB <- getPriors.NB(data1, samplesize=1000, estimation="QL", cl=NULL)
out <- getLikelihoods.NB(data1P.NB, pET="BIC", cl=NULL)
out@estProps
stat_bayseq <- out@posteriors[,2]
rank_bayseq <- rank(-stat_bayseq, ties.method="min")
datalog <- log2(data + 1)
stat_AD <- AD(data=datalog, data.cl=data.cl)
rank_AD <- rank(-abs(stat_AD), ties.method="min")
obj <- is.element(rownames(data), param3)
obj[obj == "TRUE"] <- 1
out1 <- rocdemo.sca(truth = obj, data =-rank_bayseq)
plot(out1, axes=F, ann=F, col=rgb(param7[1], param7[2], param7[3], max=255))
par(new=T)
out2 <- rocdemo.sca(truth = obj, data =-rank_AD)
plot(out2, xlab=param4, ylab=param5, main=param6, col=rgb(param8[1], param8[2], param8[3], max=255))
legend(0.6, 0.3,
c(param9, param10),
col=c(rgb(param7[1], param7[2], param7[3], max=255),
rgb(param8[1], param8[2], param8[3], max=255)
),
lwd=1,
merge=TRUE
)
AUC(out1)
AUC(out2)
5. 「4」と同じ結果だがパラメータの指定法が違う場合(多数の方法を一度に描画するときに便利です):
in_f <- "SupplementaryTable2_changed.txt"
param_G1 <- 5
param_G2 <- 5
param3 <- c("ENSG00000004468","ENSG00000182325","ENSG00000110492","ENSG00000008516","ENSG00000100170","ENSG00000173698","ENSG00000171433")
param4 <- "False Positive Rate (FPR)"
param5 <- "True Positive Rate (TPR)"
param6 <- "ROC curves for raw count data"
param7 <- list("baySeq", c( 0, 0, 0))
param8 <- list("AD", c(255, 0, 0))
source("http://www.iu.a.u-tokyo.ac.jp/~kadota/R/R_functions.R")
library(ROC)
library(baySeq)
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
groups <- list(NDE=rep(1, (param_G1+param_G2)), DE=data.cl)
data1 <- new("countData", data=as.matrix(data), replicates=data.cl, libsizes=as.integer(colSums(data)), groups=groups)
data1P.NB <- getPriors.NB(data1, samplesize=1000, estimation="QL", cl=NULL)
out <- getLikelihoods.NB(data1P.NB, pET="BIC", cl=NULL)
out@estProps
stat_bayseq <- out@posteriors[,2]
rank_bayseq <- rank(-stat_bayseq, ties.method="min")
datalog <- log2(data + 1)
stat_AD <- AD(data=datalog, data.cl=data.cl)
rank_AD <- rank(-abs(stat_AD), ties.method="min")
obj <- is.element(rownames(data), param3)
obj[obj == "TRUE"] <- 1
out1 <- rocdemo.sca(truth = obj, data =-rank_bayseq)
plot(out1, axes=F, ann=F, col=rgb(param7[[2]][1], param7[[2]][2], param7[[2]][3], max=255))
par(new=T)
out2 <- rocdemo.sca(truth = obj, data =-rank_AD)
plot(out2, xlab=param4, ylab=param5, main=param6, col=rgb(param8[[2]][1], param8[[2]][2], param8[[2]][3], max=255))
legend(0.6, 0.3,
c(param7[[1]], param8[[1]]),
col=c(rgb(param7[[2]][1], param7[[2]][2], param7[[2]][3], max=255),
rgb(param8[[2]][1], param8[[2]][2], param8[[2]][3], max=255)
),
lwd=1,
merge=TRUE
)
AUC(out1)
AUC(out2)
選択的スプライシング(Alternative Splicing; AS)はグラフで表現可能です(Sammeth, 2009)。
SplicingGraphsというパッケージはそれをうまく作成できるようです。
「ファイル」−「ディレクトリの変更」で解析したいファイルを置いてあるディレクトリに移動し以下をコピペ。
パイプライン | について
ここの項目では、公共DBからのRNA-seqデータ取得から、マッピング、カウントデータ取得、発現変動解析までの一連のコマンドを示します。
2013年秋頃にRのみで一通り行えるようにしたので記述内容を大幅に変更しています。
パイプライン | ゲノム | 発現変動 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)
Neyret-Kahn et al., Genome Res., 2013の2群間比較用ヒトRNA-seqデータ (3 proliferative samples vs. 3 Ras samples)が
GSE42213に登録されています。
ここでは、SRAdbパッケージを用いたそのFASTQ形式ファイルのダウンロードから、
QuasRパッケージを用いたマッピングおよびカウントデータ取得、
そしてTCCパッケージを用いた発現変動遺伝子(DEG)検出までを行う一連の手順を示します。
原著論文(Neyret-Kahn et al., Genome Res., 2013)では72-baseと書いてますが、取得ファイルは54-baseしかありません。
また、ヒトサンプルなのになぜかマウスゲノム("mm9")にマップしたと書いているのも意味不明です。
ちなみ54 bpと比較的長いリードであり、原著論文中でもsplice-aware alignerの一つであるTopHat (Trapnell et al., Bioinformatics, 2009)を用いてマッピングを行ったと記述していますが、
ここでは、(計算時間短縮のため)basic alignerの一つであるBowtieをQuasRの内部で用いています。
多数のファイルが作成されるので、ここでは「デスクトップ」上に「SRP017142」というフォルダを作成しておき、そこで作業を行うことにします。
Step1. RNA-seqデータのgzip圧縮済みのFASTQファイルをダウンロード:
論文中の記述からGSE42213を頼りに、
RNA-seqデータがGSE42212として収められていることを見出し、
その情報からSRP017142にたどり着いています。
したがって、ここで指定するのは"SRP017142"となります。
計6ファイル、合計6Gb程度の容量のファイルがダウンロードされます。東大の有線LANで一時間弱程度かかります。
早く終わらせたい場合は、最後のgetFASTQfile関数のオプションを'ftp'から'fasp'に変更すると時間短縮可能です。
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の記述内容と基本的に同じです。
param <- "SRP017142"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run)
getFASTQfile(hoge$run, srcType='ftp')
無事ダウンロードが終了すると、作業ディレクトリ(「デスクトップ」上の「SRP017142」フォルダ)中に7つのファイルが存在するはずです。
4Gb程度ある"SRAmetadb.sqlite"ファイルは無視して構いません。残りの"SRR"からはじまる6つのファイルがダウンロードしたRNA-seqデータです。
オリジナルのサンプル名(の略称)で対応関係を表すとsrp017142_samplename.txtのようになっていることがわかります。
尚このファイルはマッピング時の入力ファイルとしても用います。
Step2. ヒトゲノムへのマッピングおよびカウントデータ取得:
マップしたいFASTQファイルリストおよびそのサンプル名を記述したsrp017142_samplename.txtを作業ディレクトリに保存したうえで、下記を実行します。
BSgenomeパッケージで利用可能なBSgenome.Hsapiens.UCSC.hg19へマッピングしています。
名前から推測できるように"UCSC"の"hg19"にマップしているのと同じです。basic alignerの一つであるBowtieを内部的に用いており、ここではマッピング時のオプションをデフォルトにしています。
原著論文中で用いられたTopHatと同じsplice-aware alignerののカテゴリに含まれるSpliceMap (Au et al., Nucleic Acids Res., 2010)
を利用したい場合は、qAlign関数実行のところでsplicedAlignmentオプションをBowtieに対応する"F"からSpliceMapに対応する"T"に変更してください。hg19にマップした結果であり、TxDbオブジェクト取得時のゲノム情報もそれを基本として
Ensembl Genes ("ensGene")を指定しているので、Ensembl Gene IDに対するカウントデータ取得になっています。
マシンパワーにもよりますが、ノートPCでも10時間程度で終わると思います。
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)の記述内容と基本的に同じです。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f1 <- "srp017142_QC_bowtie.pdf"
out_f2 <- "srp017142_count_bowtie.txt"
out_f3 <- "srp017142_genelength.txt"
out_f4 <- "srp017142_RPKM_bowtie.txt"
out_f5 <- "srp017142_transcript_seq.fa"
out_f6 <- "srp017142_other_info1.txt"
param1 <- "hg19"
param2 <- "ensGene"
param3 <- "gene"
library(QuasR)
library(GenomicFeatures)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2,
splicedAlignment=F)
time_e <- proc.time()
qQCReport(out, pdfFilename=out_f1)
palette("default")
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
count <- qCount(out, txdb, reportLevel=param3)
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
nf_RPM <- 1000000/colSums(data)
RPM <- sweep(data, 2, nf_RPM, "*")
nf_RPK <- 1000/genelength
RPKM <- sweep(RPM, 1, nf_RPK, "*")
tmp <- cbind(rownames(RPKM), RPKM)
write.table(tmp, out_f4, sep="\t", append=F, quote=F, row.names=F)
library(in_f2, character.only=T)
tmp <- ls(paste("package", in_f2, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- extractTranscriptSeqs(genome, txdb)
fasta
writeXStringSet(fasta, file=out_f5, format="fasta", width=50)
sink(out_f6)
cat("1. Computation time for mapping (in second).\n")
time_e - time_s
cat("\n\n2. Options used for mapping.\n")
out@alignmentParameter
cat("\n\n3. Alignment statistics.\n")
alignmentStats(out)
cat("\n\n4. Gene annotation info.\n")
txdb
cat("\n\n5. Session info.\n")
sessionInfo()
sink()
無事マッピングが終了すると、指定した6つのファイルが生成されているはずです。
- QCレポートファイル(srp017142_QC_bowtie.pdf):Quality Controlレポートです。よく利用されるFastQCのようなものです。
- カウントデータファイル(srp017142_count_bowtie.txt):グループ(サンプル)間での発現変動遺伝子同定に用います。
- 遺伝子配列長情報ファイル(srp017142_genelength.txt):配列長とカウント数の関係を調べたいときなどに用います。これはおまけです。
- RPKM補正後のファイル(srp017142_RPKM_bowtie.txt):同一サンプル内での発現レベルの大小関係を知りたいときなどに用います。
- 転写物塩基配列ファイル(srp017142_transcript_seq.fa):(遺伝子ではなく)転写物の塩基配列のmulti-FASTAファイルです。参考まで。
- その他の各種情報ファイル(srp017142_other_info1.txt):論文作成時に必要な、マッピング時に用いたオプション情報、マップされたリード数、Rおよび用いたパッケージのバージョン情報などを含みます。
Step3. サンプル間クラスタリング:
カウントデータ(srp017142_count_bowtie.txt)を用いてサンプル間の全体的な類似度を眺めることを目的として、
サンプル間クラスタリングを行います。類似度は「1-Spearman相関係数」、方法は平均連結法で行っています。
TCC論文(Sun et al., 2013)のFig.3でも同じ枠組みでクラスタリングを行った結果を示していますので、英語論文執筆時の参考にどうぞ。
PearsonではなくSpearmanで行っているのは、ダイナミックレンジが広いので、順序尺度程度にしておいたほうがいいだろうという思想が一番大きいです。
log2変換してダイナミックレンジを圧縮してPearsonにするのも一般的には「アリ」だとは思いますが、マップされたリード数が100万以上あるにも関わらずRPKMデータを用いると、RPKM補正後の値が1未満のものがかなり存在すること、
そしてlogをとれるようにゼロカウントデータの処理が必要ですがやりかた次第で結果がころころかわりうるという状況が嫌なので、RNA-seqデータの場合には私はSpearman相関係数にしています。
また、ベクトルの要素間の差を基本とするdistance metrics (例:ユークリッド距離やマンハッタン距離など)は、比較的最近のRNA-seqデータ正規化法
(TMM: Robinson and Oshlack, 2010, TbT: Kadota et al., 2012, TCC; Sun et al., 2013)論文の重要性が理解できれば、その類似度は少なくともfirst choiceにならないと思われます。
つまり、サンプルごとに転写物の組成比が異なるため、RPMやCPMのような総リード数を補正しただけのデータを用いて「サンプル間の数値の差」に基づいて距離を定めるのはいかがなものか?という思想です。
逆に、ユークリッド距離などを用いてクラスタリングを行った結果と比較することで、転写物の組成比に関する知見が得られるのかもしれません。
さらに、全体的な発現レベルが低いものを予めフィルタリングしておく必要もあるのだろうとは思います。このあたりは、真の回答はありませんので、
(手持ちのデータにこの類似度を適用したときの理論上の短所をきちんと理解したうえで)いろいろ試すというのは重要だとは思います。
ここではカウントデータでクラスタリングをしていますが、おそらく配列長補正後のRPKMデータ(srp017142_RPKM_bowtie.txt)
でも得られる樹形図のトポロジー(相対的な位置関係)はほぼ同じになるのではないかと思っています。配列長補正の有無で、サンプル間の相関係数の値自体は変わりますが、
同じグループに属するサンプルであれば反復実験間でそれほど違わないので、多少順位に変動があっても全体としては相殺されるはずです...が確証はありません。
in_f3 <- "srp017142_count_bowtie.txt"
out_f6 <- "srp017142_count_cluster.png"
param_fig <- c(500, 400)
data <- read.table(in_f3, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data.dist <- as.dist(1 - cor(data, method="spearman"))
out <- hclust(data.dist, method="average")
png(out_f6, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
無事計算が終了すると、指定したファイル(srp017142_count_cluster.png)が生成されているはずです。
Step4. 発現変動遺伝子(DEG)同定:
カウントデータファイル(srp017142_count_bowtie.txt)を入力として2群間で発現の異なる遺伝子の検出を行います。
このデータはbiological replicatesありのデータなので、TCCパッケージ(Sun et al., 2013)の推奨ガイドラインに従って、
iDEGES/edgeR正規化(Sun et al., 2013; Robinson et al., 2010; Robinson and Oshlack, 2010; Robinson and Smyth, 2008)を行ったのち、
edgeRパッケージ中のan exact test (Robinson and Smyth, 2008)を行って、DEG検出を行っています。
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | TCC(Sun_2013)および
正規化 | サンプル間 | 2群間 | 複製あり | iDEGES/edgeR(Sun_2013)の記述内容と基本的に同じです。
in_f4 <- "srp017142_count_bowtie.txt"
out_f7 <- "srp017142_DEG_bowtie.txt"
out_f8 <- "srp017142_MAplot_bowtie.png"
out_f9 <- "srp017142_other_info2.txt"
param_G1 <- 3
param_G2 <- 3
param_FDR <- 0.05
param_fig <- c(430, 390)
library(TCC)
data <- read.table(in_f4, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(tcc$count), normalized, result)
write.table(tmp, out_f7, sep="\t", append=F, quote=F, row.names=F)
png(out_f8, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sink(out_f9)
cat("1. Numbers of DEGs satisfying several FDR thresholds.\n")
cat("FDR < 0.05:");print(sum(tcc$stat$q.value < 0.05))
cat("FDR < 0.10:");print(sum(tcc$stat$q.value < 0.10))
cat("FDR < 0.20:");print(sum(tcc$stat$q.value < 0.20))
cat("FDR < 0.30:");print(sum(tcc$stat$q.value < 0.30))
cat("\n\n2. Session info.\n")
sessionInfo()
sink()
無事計算が終了すると、指定した3つのファイルが生成されているはずです。
- 発現変動解析結果ファイル(srp017142_DEG_bowtie.txt):iDEGES/edgeR-edgeRパイプラインによるDEG同定結果です。
"rank"列でソートすると発現変動の度合い順になります。"q.value"列の情報は任意のFDR閾値を満たす遺伝子数を調べるときに用います。
尚、左側の実数の数値データはiDEGES/edgeR正規化後のデータです。M-A plotはこの数値データに基づいて作成されています。尚、配列長補正は掛かっておりませんのでご注意ください。
- M-A plotファイル(srp017142_MAplot_bowtie.png):M versus A plotです。横軸が平均発現レベル(右側が高発現、左側が低発現)。縦軸がlog(G2/G1)で、0より下がG1群で高発現、0より上がG2群で高発現です。
- その他の各種情報ファイル(srp017142_other_info2.txt):FDR < 0.05, 0.1, 0.2, 0.3を満たす遺伝子数、論文作成時に必要な、Rおよび用いたパッケージのバージョン情報(特にTCC)などを含みます。
Step5. iDEGES/edgeR正規化後のデータに配列長補正を実行:
カウントデータファイル(srp017142_count_bowtie.txt)と遺伝子配列長情報ファイル(srp017142_genelength.txt)を入力として
TCCパッケージ(Sun et al., 2013)の推奨ガイドラインに従って、
iDEGES/edgeR正規化(Sun et al., 2013; Robinson et al., 2010; Robinson and Oshlack, 2010; Robinson and Smyth, 2008)を行ったのち、
配列長補正(Reads per kilobase (RPK) or Counts per kilobase (CPK))を実行した結果を返します。
そろそろ誰かが論文で公式に言い出すかもしれません(既にどこかで書かれているかも...)が、RPM (Reads per million)が提唱されたのは、総リード数が100万程度だった時代です。
今はマップされた総リード数が数千万リードという時代ですので、RPM or RPKMのような100万にするような補正後のデータだと、
(細かい点をすっとばすと)せっかく読んだ総リード数の桁が増えてもダイナミックレンジが広くなりようがありません。
iDEGES/edgeR正規化後のデータは、マップされた総リード数の中央値(median)に合わせているので、リード数が増えるほどダイナミックレンジは広くなります。
しかし、これ自体は配列長補正がかかっていないため、RPKMデータと似たような「配列長補正まで行った後のiDEGES/edgeR正規化データ」があったほうが嬉しいヒトがいるのかな、ということで提供しています。
利用法としては、サンプル間クラスタリングを行う際に、順位相関係数以外のサンプルベクトル中の要素間の差に基づくユークリッド距離やマンハッタン距離をどうしても使いたい場合には
RPKMのようなデータを使うよりはこちらの正規化データのほうがnon-DEG間の距離がより0に近い値になるから直感的にはいいのではと思っています。
ただし、iDEGES/edgeR正規化を行うときにサンプルのラベル情報を用いていながら(supervised learningみたいなことを行っている)、
unsupervised learningの一種であるクラスタリングを行う、ということの妥当性についてはよくわかりません。
正規化 | サンプル間 | 2群間 | 複製あり | iDEGES/edgeR(Sun_2013)と
正規化 | 基礎 | RPK or CPK (配列長補正)の記述内容と基本的に同じです。
in_f4 <- "srp017142_count_bowtie.txt"
in_f5 <- "srp017142_genelength.txt"
out_f10 <- "srp017142_normalized_bowtie.txt"
param_G1 <- 3
param_G2 <- 3
library(TCC)
data <- read.table(in_f4, header=TRUE, row.names=1, sep="\t", quote="")
len <- read.table(in_f5, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
normalized <- getNormalizedData(tcc)
nf <- 1000/len[,1]
normalizedK <- sweep(normalized, 1, nf, "*")
tmp <- cbind(rownames(tcc$count), normalizedK)
write.table(tmp, out_f10, sep="\t", append=F, quote=F, row.names=F)
無事計算が終了すると、配列長補正まで行った後のiDEGES/edgeR正規化データファイル(srp017142_normalized_bowtie.txt)が生成されているはずです。
- SRP017142:Neyret-Kahn et al., Genome Res., 2013
- SRAdb:Zhu et al., BMC Bioinformatics, 2013
- QuasR:Gaidatzis et al., Bioinformatics, 2015
- TCC:Sun et al., BMC Bioinformatics, 2013
- Bowtie:Langmead et al., Genome Biol., 2009
- SpliceMap:Au et al., Nucleic Acids Res., 2010
- GenomicFeatures:Lawrence et al., PLoS Comput. Biol., 2013
- edgeR:Robinson et al., Bioinformatics, 2010
- TMM正規化法:Robinson and Oshlack, Genome Biol., 2010
- an exact test for negative binomial distribution:Robinson and Smyth, Biostatistics, 2008
パイプライン | ゲノム | 機能解析 | 2群間 | 対応なし | 複製あり | SRP017142(Neyret-Kahn_2013)
Neyret-Kahn et al., Genome Res., 2013の2群間比較用ヒトRNA-seqデータ (3 proliferative samples vs. 3 Ras samples)が
GSE42213に登録されています。
ここでは、ファイルのダウンロードから、マッピング、カウントデータ取得、機能解析までを行う一連の手順を示します。
多数のファイルが作成されるので、ここでは「デスクトップ」上に「SRP017142」というフォルダを作成しておき、そこで作業を行うことにします。
Step1. RNA-seqデータのgzip圧縮済みのFASTQファイルをダウンロード:
論文中の記述からGSE42213を頼りに、
RNA-seqデータがGSE42212として収められていることを見出し、
その情報からSRP017142にたどり着いています。
したがって、ここで指定するのは"SRP017142"となります。
計6ファイル、合計6Gb程度の容量のファイルがダウンロードされます。東大の有線LANで一時間弱程度かかります。
早く終わらせたい場合は、最後のgetFASTQfile関数のオプションを'ftp'から'fasp'に変更すると時間短縮可能です。
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の記述内容と基本的に同じです。
param <- "SRP017142"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run)
getFASTQfile(hoge$run, srcType='ftp')
無事ダウンロードが終了すると、作業ディレクトリ(「デスクトップ」上の「SRP017142」フォルダ)中に7つのファイルが存在するはずです。
4Gb程度ある"SRAmetadb.sqlite"ファイルは無視して構いません。残りの"SRR"からはじまる6つのファイルがダウンロードしたRNA-seqデータです。
オリジナルのサンプル名(の略称)で対応関係を表すとsrp017142_samplename.txtのようになっていることがわかります。
尚このファイルはマッピング時の入力ファイルとしても用います。
Step2. ヒトゲノムへのマッピングおよびカウントデータ取得:
マップしたいFASTQファイルリストおよびそのサンプル名を記述したsrp017142_samplename.txtを作業ディレクトリに保存したうえで、下記を実行します。
BSgenomeパッケージで利用可能なBSgenome.Hsapiens.UCSC.hg19へマッピングしています。
名前から推測できるように"UCSC"の"hg19"にマップしているのと同じです。
basic alignerの一つであるBowtieを内部的に用いており、
ここではマッピング時のオプションを"-m 1 --best --strata -v 2"にしています。
hg19にマップした結果なので、TxDbオブジェクト取得時のゲノム情報もそれを基本として
Ensembl Genes ("ensGene")を指定しているので、Ensembl Gene IDに対するカウントデータ取得になっています。
但し、機能解析で用いるSeqGSEAパッケージの入力に合わせて、
exonレベルのカウントデータとして取得しています。マシンパワーにもよりますが、ノートPCでも10時間程度で終わると思います。
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)の記述内容と基本的に同じです。
in_f1 <- "srp017142_samplename.txt"
in_f2 <- "BSgenome.Hsapiens.UCSC.hg19"
out_f1 <- "srp017142_QC_bowtie2.pdf"
out_f2 <- "srp017142_count_bowtie2.txt"
out_f3 <- "srp017142_genelength2.txt"
out_f4 <- "srp017142_RPKM_bowtie2.txt"
out_f5 <- "srp017142_transcript_seq2.fa"
out_f6 <- "srp017142_other_info1_2.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param1 <- "hg19"
param2 <- "ensGene"
param3 <- "exon"
library(QuasR)
library(GenomicFeatures)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping)
time_e <- proc.time()
qQCReport(out, pdfFilename=out_f1)
palette("default")
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
count <- qCount(out, txdb, reportLevel=param3)
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
nf_RPM <- 1000000/colSums(data)
RPM <- sweep(data, 2, nf_RPM, "*")
nf_RPK <- 1000/genelength
RPKM <- sweep(RPM, 1, nf_RPK, "*")
tmp <- cbind(rownames(RPKM), RPKM)
write.table(tmp, out_f4, sep="\t", append=F, quote=F, row.names=F)
library(in_f2, character.only=T)
tmp <- ls(paste("package", in_f2, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- extractTranscriptSeqs(genome, txdb)
fasta
writeXStringSet(fasta, file=out_f5, format="fasta", width=50)
sink(out_f6)
cat("1. Computation time for mapping (in second).\n")
time_e - time_s
cat("\n\n2. Options used for mapping.\n")
out@alignmentParameter
cat("\n\n3. Alignment statistics.\n")
alignmentStats(out)
cat("\n\n4. Gene annotation info.\n")
txdb
cat("\n\n5. Session info.\n")
sessionInfo()
sink()
無事マッピングが終了すると、指定した6つのファイルが生成されているはずです。
- QCレポートファイル(srp017142_QC_bowtie2.pdf):Quality Controlレポートです。よく利用されるFastQCのようなものです。
- カウントデータファイル(srp017142_count_bowtie2.txt):グループ(サンプル)間での発現変動遺伝子同定に用います。
- 遺伝子配列長情報ファイル(srp017142_genelength2.txt):配列長とカウント数の関係を調べたいときなどに用います。これはおまけです。
- RPKM補正後のファイル(srp017142_RPKM_bowtie2.txt):同一サンプル内での発現レベルの大小関係を知りたいときなどに用います。
- 転写物塩基配列ファイル(srp017142_transcript_seq2.fa):(遺伝子ではなく)転写物の塩基配列のmulti-FASTAファイルです。参考まで。
- その他の各種情報ファイル(srp017142_other_info1_2.txt):論文作成時に必要な、マッピング時に用いたオプション情報、マップされたリード数、Rおよび用いたパッケージのバージョン情報などを含みます。
Step3. 機能解析:
カウントデータファイル(srp017142_count_bowtie2.txt)を入力として
SeqGSEAを用いて機能解析を行います。
このパッケージは基本的にexonレベルのカウントデータを入力とするので、その前提で行っています。
また、統計的有意性の評価にサンプルラベル情報の並べ替え(permutation)戦略を採用しているため、
各グループあたりの反復数が5以上のデータを想定しているようです。また、計算時間を半端なく要します(10時間とか)。
それゆえ、本来このデータセットはグループあたりの反復数が3しかないので、適用外ということになります。
Step2に引き続いて行う場合には、dataとtxdbオブジェクトの構築を行う必要は本来ありません。
同様の理由でtxdbオブジェクトの作成に必要なparam1とparam2も必要ありませんが、
これらのアノテーション情報を利用していることが既知であるという前提のもとで行っています。
最終的に欲しいReadCountSetクラスオブジェクトは、「カウントデータ、gene ID、exon ID」の3つの情報から構築されます。
しかし、txdbオブジェクトから得られるgene IDとexon IDはshared exonを含むので、曖昧性を排除するためこれらを除去する必要があります。
それゆえ、hoge2中のexon IDを抽出して一回しか出現しなかったIDをhoge4として取得し、
もとのhoge2オブジェクトの並びで取得しなおしたものがexonIDsとgeneIDsオブジェクトです。
exonレベルカウントデータもshared exonの情報を含むので、それらを除いたものを取得しています。
SeqGSEAでの機能解析の基本はexonレベルとgeneレベルの発現変動解析結果を組み合わせてGSEAを行うというものです(Wang and Cairns, 2013)。
SeqGSEA著者たちは、exonレベルの発現変動解析のことをDifferential splicing (DS) analysisと呼んでいて、
おそらくDSGseqはSeqGSEA中に組み込まれていると思います。
そしてgeneレベルの発現変動解析をdifferential expression (DE) analysisとして、SeqGSEA中では
DESeqを利用しています。
GSEAに代表される発現変動遺伝子セット解析は、基本的にGSEAの開発者らが作成した様々な遺伝子セット情報を収めたMolecular Signatures Database (MSigDB)からダウンロードした.gmt形式ファイルを読み込んで解析を行います。
*gmt形式ファイルのダウンロード方法は、基本的に以下の通りです:
- Molecular Signatures Database (MSigDB)の
「register」のページで登録し、遺伝子セットをダウンロード可能な状態にする。
- Molecular Signatures Database (MSigDB)の
「Download gene sets」の"Download"のところをクリックし、Loginページで登録したe-mail addressを入力。
- これでMSigDBのダウンロードページに行けるので、
「c5: gene ontology gene sets」の「BP: biological process」を解析したい場合はc5.bp.v4.0.symbols.gmtファイルをダウンロードしておく。
「c2: curated gene sets」の「KEGG gene sets」を解析したい場合はc2.cp.kegg.v4.0.symbols.gmtファイルをダウンロードしておく。
「c3: motif gene sets」を解析したい場合はc3.all.v4.0.symbols.gmtファイルをダウンロードしておく。
以下ではc5.bp.v4.0.symbols.gmtの解析を行っています。
また、並べ替え回数がたったの20回でも2時間ちょっとかかります(Panasonic Let's note CF-SX3本郷モデルの場合)のでご注意ください。
推奨は1000回以上と書いてますが、個人的にはアリエナイですね。。。
in_f4 <- "srp017142_count_bowtie2.txt"
in_f5 <- "c5.bp.v4.0.symbols.gmt"
out_f7 <- "srp017142_SeqGSEA_c5bp.txt"
param_G1 <- 3
param_G2 <- 3
param_perm <- 20
param1 <- "hg19"
param2 <- "ensGene"
library(SeqGSEA)
library(GenomicFeatures)
data <- read.table(in_f4, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
tmp_colname <- colnames(data)
colnames(data) <- c(paste("E", 1:param_G1, sep=""), paste("C", 1:param_G2, sep=""))
txdb <- makeTxDbFromUCSC(genome=param1, tablename=param2)
hoge1 <- exonsBy(txdb, by=c("gene"))
hoge2 <- unlist(hoge1)
hoge2
hoge3 <- table(hoge2$exon_id)
hoge4 <- names(hoge3)[hoge3 == 1]
obj <- is.element(as.character(hoge2$exon_id), hoge4)
exonIDs <- as.character(hoge2$exon_id)[obj]
geneIDs <- names(hoge2)[obj]
data <- data[exonIDs,]
#rownames(data) <- paste(geneIDs, exonIDs, sep=":")
dim(data)
exonIDs <- paste("E", exonIDs, sep="")
RCS <- newReadCountSet(data, exonIDs, geneIDs)
RCS
RCS <- exonTestability(RCS, cutoff = 5)
geneTestable <- geneTestability(RCS)
RCS <- subsetByGenes(RCS, unique(geneID(RCS))[geneTestable])
RCS
time_DS_s <- proc.time()
RCS <- estiExonNBstat(RCS)
RCS <- estiGeneNBstat(RCS)
head(fData(RCS)[, c("exonIDs", "geneIDs", "testable", "NBstat")])
permuteMat <- genpermuteMat(RCS, times=param_perm)
RCS <- DSpermute4GSEA(RCS, permuteMat)
DSscore.normFac <- normFactor(RCS@permute_NBstat_gene)
DSscore <- scoreNormalization(RCS@featureData_gene$NBstat, DSscore.normFac)
DSscore.perm <- scoreNormalization(RCS@permute_NBstat_gene, DSscore.normFac)
RCS <- DSpermutePval(RCS, permuteMat)
head(DSresultGeneTable(RCS))
time_DS_e <- proc.time()
time_DS_e - time_DS_s
time_DE_s <- proc.time()
geneCounts <- getGeneCount(RCS)
head(geneCounts)
DEG <- runDESeq(geneCounts, label(RCS))
DEGres <- DENBStat4GSEA(DEG)
DEpermNBstat <- DENBStatPermut4GSEA(DEG, permuteMat)
DEscore.normFac <- normFactor(DEpermNBstat)
DEscore <- scoreNormalization(DEGres$NBstat, DEscore.normFac)
DEscore.perm <- scoreNormalization(DEpermNBstat, DEscore.normFac)
DEGres <- DEpermutePval(DEGres, DEpermNBstat)
head(DEGres)
time_DE_e <- proc.time()
time_DE_e - time_DE_s
combine <- rankCombine(DEscore, DSscore, DEscore.perm, DSscore.perm, DEweight = 0.3)
gene.score <- combine$geneScore
gene.score.perm <- combine$genePermuteScore
GS <- loadGenesets(in_f5, unique(geneID(RCS)), geneID.type = "ensembl")
GS <- GSEnrichAnalyze(GS, gene.score, gene.score.perm)
#tmp <- GSEAresultTable(GS, GSDesc = TRUE)
tmp <- topGeneSets(GS, n=length(GS@GSNames), sortBy="FDR")
write.table(tmp, out_f7, sep="\t", append=F, quote=F, row.names=F)
無事計算が終了すると、指定したファイル(srp017142_SeqGSEA_c5bp.txt)が生成されているはずです。
このファイルは並べ替え回数を30回(param_perm <- 30)にして実行した結果です。
permutation p-valueに基づく結果であり並べ替え回数が少ないので、同じ数値を指定した計算結果でも、全く同じ数値や並びになっているとは限りませんのでご注意ください。
例えば、このファイルの場合は、FDR < 0.05未満のものが22個(遺伝子セット名が"CYTOKINESIS"から"INNATE_IMMUNE_RESPONSE"まで)あると解釈します。
- SRP017142:Neyret-Kahn et al., Genome Res., 2013
- DDBJ Sequence Read Archive (DRA):Kodama et al., Nucleic Acids Res., 2012
- QuasR:Gaidatzis et al., Bioinformatics, 2015
- Bowtie:Langmead et al., Genome Biol., 2009
- GenomicFeatures:Lawrence et al., PLoS Comput. Biol., 2013
- SeqGSEA(パッケージの論文):Wang et al., Bioinformatics, 2014
- SeqGSEA(の原著論文):Wang and Cairns, BMC Bioinformatics, 2013
- DSGseq:Wang et al., Gene, 2013
- Molecular Signatures Database (MSigDB):Subramanian et al., PNAS, 2005
パイプライン | ゲノム | 機能解析 | 2群間 | 対応なし | 複製あり | SRP011435(Huang_2012)
Huang et al., Development, 2012の2群間比較用シロイヌナズナRNA-seqデータ (4 DEX-treated vs. 4 mock-treated)が
GSE36469に登録されています。
ここでは、SRAdbパッケージを用いたそのFASTQ形式ファイルのダウンロードから、
QuasRパッケージを用いたマッピングおよびカウントデータ取得、
そしてTCCパッケージを用いた発現変動遺伝子(DEG)検出までを行う一連の手順を示します。
多数のファイルが作成されるので、ここでは「デスクトップ」上に「SRP011435」というフォルダを作成しておき、そこで作業を行うことにします。
Step1. RNA-seqデータのgzip圧縮済みのFASTQファイルをダウンロード:
論文中の記述からGSE36469を頼りに、
RNA-seqデータがGSE36469として収められていることを見出し、
その情報からSRP011435にたどり着いています。
したがって、ここで指定するのは"SRP011435"となります。
計8ファイル、合計10Gb程度の容量のファイルがダウンロードされます。東大の有線LANで2時間程度かかります。
早く終わらせたい場合は、最後のgetFASTQfile関数のオプションを'ftp'から'fasp'に変更すると時間短縮可能です。
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の記述内容と基本的に同じです。
param <- "SRP011435"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run)
getFASTQfile(hoge$run, srcType='ftp')
無事ダウンロードが終了すると、作業ディレクトリ(「デスクトップ」上の「SRP011435」フォルダ)中に9つのファイルが存在するはずです。
4Gb程度ある"SRAmetadb.sqlite"ファイルは無視して構いません。残りの"SRR"からはじまる8つのファイルがダウンロードしたRNA-seqデータです。
オリジナルのサンプル名(の略称)で対応関係を表すとsrp011435_samplename.txtのようになっていることがわかります。
尚このファイルはマッピング時の入力ファイルとしても用います。
Step2. シロイヌナズナ(A. thaliana)ゲノムへのマッピングおよびカウントデータ取得:
マップしたいFASTQファイルリストおよびそのサンプル名を記述したsrp011435_samplename.txtを作業ディレクトリに保存したうえで、下記を実行します。
BSgenomeパッケージで利用可能なBSgenome.Athaliana.TAIR.TAIR9へマッピングしています。
名前から推測できるように"TAIR"の"TAIR9"にマップしているのと同じです。BSgenome.Athaliana.TAIR.TAIR9パッケージインストールされていない場合は自動でインストールしてくれるので特に気にする必要はありません。
basic alignerの一つであるBowtieを内部的に用いており、ここではマッピング時のオプションをデフォルトにしています。
原著論文中で用いられたTopHatと同じsplice-aware alignerののカテゴリに含まれるSpliceMap (Au et al., Nucleic Acids Res., 2010)
を利用したい場合は、qAlign関数実行のところでsplicedAlignmentオプションをBowtieに対応する"F"からSpliceMapに対応する"T"に変更してください。
TAIR9にマップした結果であり、UCSCからはArabidopsisの遺伝子アノテーション情報が提供されていないため、
TAIR10_GFF3_genes.gffを予めダウンロードしておき、makeTxDbFromGFF関数を用いてTxDbオブジェクトを作成しています。
マシンパワーにもよりますが、ノートPCでも10時間程度で終わると思います。
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)の記述内容と基本的に同じです。
in_f1 <- "srp011435_samplename.txt"
in_f2 <- "BSgenome.Athaliana.TAIR.TAIR9"
in_f3 <- "TAIR10_GFF3_genes.gff"
out_f1 <- "srp011435_QC_bowtie.pdf"
out_f2 <- "srp011435_count_bowtie.txt"
out_f3 <- "srp011435_genelength.txt"
out_f4 <- "srp011435_RPKM_bowtie.txt"
out_f5 <- "srp011435_transcript_seq.fa"
out_f6 <- "srp011435_other_info1.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param3 <- "gene"
library(QuasR)
library(GenomicFeatures)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping,
splicedAlignment=F)
time_e <- proc.time()
qQCReport(out, pdfFilename=out_f1)
palette("default")
time_e - time_s
txdb <- makeTxDbFromGFF(in_f3)
count <- qCount(out, txdb, reportLevel=param3)
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
nf_RPM <- 1000000/colSums(data)
RPM <- sweep(data, 2, nf_RPM, "*")
nf_RPK <- 1000/genelength
RPKM <- sweep(RPM, 1, nf_RPK, "*")
tmp <- cbind(rownames(RPKM), RPKM)
write.table(tmp, out_f4, sep="\t", append=F, quote=F, row.names=F)
library(in_f2, character.only=T)
tmp <- ls(paste("package", in_f2, sep=":"))
genome <- eval(parse(text=tmp))
fasta <- extractTranscriptSeqs(genome, txdb)
fasta
writeXStringSet(fasta, file=out_f5, format="fasta", width=50)
sink(out_f6)
cat("1. Computation time for mapping (in second).\n")
time_e - time_s
cat("\n\n2. Options used for mapping.\n")
out@alignmentParameter
cat("\n\n3. Alignment statistics.\n")
alignmentStats(out)
cat("\n\n4. Gene annotation info.\n")
txdb
cat("\n\n5. Session info.\n")
sessionInfo()
sink()
無事マッピングが終了すると、指定した6つのファイルが生成されているはずです。
- QCレポートファイル(srp011435_QC_bowtie.pdf):Quality Controlレポートです。よく利用されるFastQCのようなものです。
- カウントデータファイル(srp011435_count_bowtie.txt):グループ(サンプル)間での発現変動遺伝子同定に用います。
- 遺伝子配列長情報ファイル(srp011435_genelength.txt):配列長とカウント数の関係を調べたいときなどに用います。これはおまけです。
- RPKM補正後のファイル(srp011435_RPKM_bowtie.txt):同一サンプル内での発現レベルの大小関係を知りたいときなどに用います。
- 転写物塩基配列ファイル(srp011435_transcript_seq.fa):(遺伝子ではなく)転写物の塩基配列のmulti-FASTAファイルです。参考まで。
- その他の各種情報ファイル(srp011435_other_info1.txt):論文作成時に必要な、マッピング時に用いたオプション情報、マップされたリード数、Rおよび用いたパッケージのバージョン情報などを含みます。
Step2. シロイヌナズナ(A. thaliana)ゲノムへのマッピングおよびカウントデータ取得(リファレンスがmulti-FASTAファイルの場合):
マップしたいFASTQファイルリストおよびそのサンプル名を記述したsrp011435_samplename.txtを作業ディレクトリに保存したうえで、下記を実行します。
シロイヌナズナのゲノム配列ファイル(TAIR10_chr_all.fas.gz)へマッピングしています。
但し、マッピングに用いるQuasRパッケージ中のqAlign関数がリファレンス配列ファイルの拡張子として"*.fasta", "*.fa", "*.fna"しか認識してくれません。
また、カウントデータを取得するために遺伝子アノテーションファイル(TAIR10_GFF3_genes.gff)を利用する必要がありますが、
このファイル中の染色体名と揃える必要があるため、TAIR10_chr_all.fas.gzファイル中のdescription部分をparamで指定した文字列に置換したファイル(tmp_genome.fasta)を中間ファイルとして作成しています。
TAIR10_GFF3_genes.gffを予めダウンロードしておき、makeTxDbFromGFF関数を用いてTxDbオブジェクトを作成しています。
マシンパワーにもよりますが、ノートPCでも10時間程度で終わると思います。マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)の記述内容と基本的に同じです。
2014年5月2日までextractTranscriptsFromGenome関数を用いて転写物配列情報を取得していましたが、「'extractTranscriptsFromGenome' is deprecated. Use 'extractTranscriptSeqs' instead.」
という警告メッセージが出たので、extractTranscriptSeqs関数に変更しています。尚、Step2のトータル計算時間はノートPCで7時間程度です。
in_f <- "TAIR10_chr_all.fas.gz"
out_f <- "tmp_genome.fasta"
param <- c("Chr1","Chr2","Chr3","Chr4","Chr5","ChrM","ChrC")
library(Biostrings)
fasta <- readDNAStringSet(in_f, format="fasta")
fasta
names(fasta) <- param
fasta
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
in_f1 <- "srp011435_samplename.txt"
in_f2 <- "tmp_genome.fasta"
in_f3 <- "TAIR10_GFF3_genes.gff"
out_f1 <- "srp011435_QC_bowtie_2.pdf"
out_f2 <- "srp011435_count_bowtie_2.txt"
out_f3 <- "srp011435_genelength_2.txt"
out_f4 <- "srp011435_RPKM_bowtie_2.txt"
#out_f5 <- "srp011435_transcript_seq_2.fa"
out_f6 <- "srp011435_other_info1_2.txt"
param_mapping <- "-m 1 --best --strata -v 2"
param3 <- "gene"
library(QuasR)
library(GenomicFeatures)
#library(Rsamtools)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2, alignmentParameter=param_mapping,
splicedAlignment=F)
time_e <- proc.time()
qQCReport(out, pdfFilename=out_f1)
palette("default")
time_e - time_s
txdb <- makeTxDbFromGFF(in_f3)
count <- qCount(out, txdb, reportLevel=param3)
data <- count[,-1]
tmp <- cbind(rownames(data), data)
write.table(tmp, out_f2, sep="\t", append=F, quote=F, row.names=F)
genelength <- count[,1]
tmp <- cbind(names(genelength), genelength)
write.table(tmp, out_f3, sep="\t", append=F, quote=F, row.names=F)
nf_RPM <- 1000000/colSums(data)
RPM <- sweep(data, 2, nf_RPM, "*")
nf_RPK <- 1000/genelength
RPKM <- sweep(RPM, 1, nf_RPK, "*")
tmp <- cbind(rownames(RPKM), RPKM)
write.table(tmp, out_f4, sep="\t", append=F, quote=F, row.names=F)
#fasta_trans <- extractTranscriptSeqs(FaFile(in_f2), txdb)
#fasta_trans <- extractTranscriptSeqs(FaFile(in_f2), exonsBy(txdb, by="tx", use.names=TRUE))
#fasta_trans <- extractTranscriptSeqs(FaFile(in_f2), cdsBy(txdb, by="tx", use.names=TRUE))
#fasta_trans <- extractTranscriptSeqs(fasta, txdb)
#fasta_trans <- extractTranscriptSeqs(fasta, exonsBy(txdb, by="tx", use.names=TRUE))
#fasta_trans
#writeXStringSet(fasta_trans, file=out_f5, format="fasta", width=50)
sink(out_f6)
cat("1. Computation time for mapping (in second).\n")
time_e - time_s
cat("\n\n2. Options used for mapping.\n")
out@alignmentParameter
cat("\n\n3. Alignment statistics.\n")
alignmentStats(out)
cat("\n\n4. Gene annotation info.\n")
txdb
cat("\n\n5. Session info.\n")
sessionInfo()
sink()
無事マッピングが終了すると、指定した6つのファイルが生成されているはずです。
- QCレポートファイル(srp011435_QC_bowtie_2.pdf):Quality Controlレポートです。よく利用されるFastQCのようなものです。
- カウントデータファイル(srp011435_count_bowtie_2.txt):グループ(サンプル)間での発現変動遺伝子同定に用います。
- 遺伝子配列長情報ファイル(srp011435_genelength_2.txt):配列長とカウント数の関係を調べたいときなどに用います。これはおまけです。
- RPKM補正後のファイル(srp011435_RPKM_bowtie_2.txt):同一サンプル内での発現レベルの大小関係を知りたいときなどに用います。
- その他の各種情報ファイル(srp011435_other_info1_2.txt):論文作成時に必要な、マッピング時に用いたオプション情報、マップされたリード数、Rおよび用いたパッケージのバージョン情報などを含みます。
Step3. サンプル間クラスタリング:
カウントデータ(srp011435_count_bowtie_2.txt)を用いてサンプル間の全体的な類似度を眺めることを目的として、サンプル間クラスタリングを行います。
類似度は「1-Spearman相関係数」、方法は平均連結法で行っています。
TCC論文(Sun et al., 2013)のFig.3でも同じ枠組みでクラスタリングを行った結果を示していますので、英語論文執筆時の参考にどうぞ。
PearsonではなくSpearmanで行っているのは、ダイナミックレンジが広いので、順序尺度程度にしておいたほうがいいだろうという思想が一番大きいです。
log2変換してダイナミックレンジを圧縮してPearsonにするのも一般的には「アリ」だとは思いますが、マップされたリード数が100万以上あるにも関わらずRPKMデータを用いると、RPKM補正後の値が1未満のものがかなり存在すること、
そしてlogをとれるようにゼロカウントデータの処理が必要ですがやりかた次第で結果がころころかわりうるという状況が嫌なので、RNA-seqデータの場合には私はSpearman相関係数にしています。
また、ベクトルの要素間の差を基本とするdistance metrics (例:ユークリッド距離やマンハッタン距離など)は、比較的最近のRNA-seqデータ正規化法
(TMM: Robinson and Oshlack, 2010, TbT: Kadota et al., 2012, TCC; Sun et al., 2013)論文の重要性が理解できれば、その類似度は少なくともfirst choiceにならないと思われます。
つまり、サンプルごとに転写物の組成比が異なるため、RPMやCPMのような総リード数を補正しただけのデータを用いて「サンプル間の数値の差」に基づいて距離を定めるのはいかがなものか?という思想です。
逆に、ユークリッド距離などを用いてクラスタリングを行った結果と比較することで、転写物の組成比に関する知見が得られるのかもしれません。
さらに、全体的な発現レベルが低いものを予めフィルタリングしておく必要もあるのだろうとは思います。このあたりは、真の回答はありませんので、
(手持ちのデータにこの類似度を適用したときの理論上の短所をきちんと理解したうえで)いろいろ試すというのは重要だとは思います。
ここではカウントデータでクラスタリングをしていますが、おそらく配列長補正後のRPKMデータ(srp011435_RPKM_bowtie_2.txt)
でも得られる樹形図のトポロジー(相対的な位置関係)はほぼ同じになるのではないかと思っています。配列長補正の有無で、サンプル間の相関係数の値自体は変わりますが、
同じグループに属するサンプルであれば反復実験間でそれほど違わないので、多少順位に変動があっても全体としては相殺されるはずです...が確証はありません。
in_f3 <- "srp011435_count_bowtie_2.txt"
out_f6 <- "srp011435_count_cluster.png"
param_fig <- c(500, 400)
data <- read.table(in_f3, header=TRUE, row.names=1, sep="\t", quote="")
dim(data)
obj <- as.logical(rowSums(data) > 0)
data <- unique(data[obj,])
dim(data)
data.dist <- as.dist(1 - cor(data, method="spearman"))
out <- hclust(data.dist, method="average")
png(out_f6, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
上と同じ結果は、TCC ver. 1.4.0以降で
clusterSample関数を用いて得ることができます。
in_f3 <- "srp011435_count_bowtie_2.txt"
out_f6 <- "hoge.png"
param_fig <- c(500, 400)
library(TCC)
data <- read.table(in_f3, header=TRUE, row.names=1, sep="\t", quote="")
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
png(out_f6, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(out)
dev.off()
無事計算が終了すると、指定したファイル(srp011435_count_cluster.png)が生成されているはずです。
Step4. 発現変動遺伝子(DEG)同定:
カウントデータファイル(srp011435_count_bowtie_2.txt)を入力として2群間で発現の異なる遺伝子の検出を行います。
このデータはtechnical replicatesを含むので、それをマージしたのちbiological replicatesのデータにしてからTCCパッケージ(Sun et al., 2013)の推奨ガイドラインに従って、
iDEGES/edgeR正規化(Sun et al., 2013; Robinson et al., 2010; Robinson and Oshlack, 2010; Robinson and Smyth, 2008)を行ったのち、
edgeRパッケージ中のan exact test (Robinson and Smyth, 2008)を行って、DEG検出を行っています。
解析 | 発現変動 | 2群間 | 対応なし | 複製あり | TCC(Sun_2013)の記述内容と基本的に同じです。
technical replicatesデータのマージ。ここでは、アドホックに2列分ごとのサブセットを抽出し、行の総和を計算したのち、結合しています。
in_f <- "srp011435_count_bowtie_2.txt"
out_f <- "srp011435_count_bowtie_3.txt"
data <- read.table(in_f, header=TRUE, row.names=1, sep="\t", quote="")
head(data)
DEX_bio1 <- rowSums(data[,1:2])
DEX_bio2 <- rowSums(data[,3:4])
mock_bio1 <- rowSums(data[,5:6])
mock_bio2 <- rowSums(data[,7:8])
out <- cbind(DEX_bio1, DEX_bio2, mock_bio1, mock_bio2)
head(out)
tmp <- cbind(rownames(out), out)
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F)
in_f4 <- "srp011435_count_bowtie_3.txt"
out_f7 <- "srp011435_DEG_bowtie.txt"
out_f8 <- "srp011435_MAplot_bowtie.png"
out_f9 <- "srp011435_other_info2.txt"
param_G1 <- 2
param_G2 <- 2
param_FDR <- 0.05
param_fig <- c(430, 390)
library(TCC)
data <- read.table(in_f4, header=TRUE, row.names=1, sep="\t", quote="")
data.cl <- c(rep(1, param_G1), rep(2, param_G2))
tcc <- new("TCC", data, data.cl)
tcc <- calcNormFactors(tcc, norm.method="tmm", test.method="edger",
iteration=3, FDR=0.1, floorPDEG=0.05)
tcc <- estimateDE(tcc, test.method="edger", FDR=param_FDR)
result <- getResult(tcc, sort=FALSE)
sum(tcc$stat$q.value < param_FDR)
normalized <- getNormalizedData(tcc)
tmp <- cbind(rownames(tcc$count), normalized, result)
write.table(tmp, out_f7, sep="\t", append=F, quote=F, row.names=F)
png(out_f8, pointsize=13, width=param_fig[1], height=param_fig[2])
plot(tcc, FDR=param_FDR)
legend("topright", c(paste("DEG(FDR<", param_FDR, ")", sep=""), "non-DEG"),
col=c("magenta", "black"), pch=20)
dev.off()
sink(out_f9)
cat("1. Numbers of DEGs satisfying several FDR thresholds.\n")
cat("FDR < 0.05:");print(sum(tcc$stat$q.value < 0.05))
cat("FDR < 0.10:");print(sum(tcc$stat$q.value < 0.10))
cat("FDR < 0.20:");print(sum(tcc$stat$q.value < 0.20))
cat("FDR < 0.30:");print(sum(tcc$stat$q.value < 0.30))
cat("\n\n2. Session info.\n")
sessionInfo()
sink()
無事計算が終了すると、指定した3つのファイルが生成されているはずです。
- 発現変動解析結果ファイル(srp011435_DEG_bowtie.txt):iDEGES/edgeR-edgeRパイプラインによるDEG同定結果です。
"rank"列でソートすると発現変動の度合い順になります。"q.value"列の情報は任意のFDR閾値を満たす遺伝子数を調べるときに用います。
尚、左側の実数の数値データはiDEGES/edgeR正規化後のデータです。M-A plotはこの数値データに基づいて作成されています。尚、配列長補正は掛かっておりませんのでご注意ください。
- M-A plotファイル(srp011435_MAplot_bowtie.png):M versus A plotです。横軸が平均発現レベル(右側が高発現、左側が低発現)。縦軸がlog(G2/G1)で、0より下がG1群で高発現、0より上がG2群で高発現です。
- その他の各種情報ファイル(srp011435_other_info2.txt):FDR < 0.05, 0.1, 0.2, 0.3を満たす遺伝子数、論文作成時に必要な、Rおよび用いたパッケージのバージョン情報(特にTCC)などを含みます。
- SRAdb:Zhu et al., BMC Bioinformatics, 2013
- SRP017142:Neyret-Kahn et al., Genome Res., 2013
- QuasR:Gaidatzis et al., Bioinformatics, 2015
- Bowtie:Langmead et al., Genome Biol., 2009
- GenomicFeatures:Lawrence et al., PLoS Comput. Biol., 2013
- TCC:Sun et al., BMC Bioinformatics, 2013
パイプライン | ゲノム | small RNA | SRP016842(Nie_2013)
Nie et al., BMC Genomics, 2013のカイコ (Bombyx mori) small RNA-seqデータが
GSE41841に登録されています。
(そしてリンク先のGSM1025527からも様々な情報を得ることができます。)
ここでは、SRAdbパッケージを用いたそのFASTQ形式ファイルのダウンロードから、
QuasRパッケージを用いたマッピングまでを行う一連の手順を示します。
basic alignerの一つであるBowtieをQuasRの内部で用いています。
ここでは「デスクトップ」上に「SRP016842」というフォルダを作成しておき、そこで作業を行うことにします。
Step1. RNA-seqデータのgzip圧縮済みのFASTQファイルをダウンロード:
論文中の記述からGSE41841を頼りに、
SRP016842にたどり着いています。
したがって、ここで指定するのは"SRP016842"となります。
以下を実行して得られるsmall RNA-seqファイルは一つ(SRR609266.fastq.gz)で、ファイルサイズは400Mb弱、11928428リードであることがわかります。
イントロ | NGS | 配列取得 | FASTQ or SRA | SRAdb(Zhu_2013)の記述内容と基本的に同じです。
param <- "SRP016842"
library(SRAdb)
#sqlfile <- "SRAmetadb.sqlite"
sqlfile <- getSRAdbFile()
sra_con <- dbConnect(SQLite(), sqlfile)
hoge <- sraConvert(param, sra_con=sra_con)
hoge
apply(hoge, 2, unique)
getFASTQinfo(in_acc=hoge$run)
getFASTQfile(hoge$run, srcType='ftp')
無事ダウンロードが終了すると、作業ディレクトリ(「デスクトップ」上の「SRP016842」フォルダ)中に2つのファイルが存在するはずです。
4Gb程度ある"SRAmetadb.sqlite"ファイルは無視して構いません。残りの"SRR"からはじまるファイル(SRR609266.fastq.gz)がダウンロードしたsRNA-seqデータです。
オリジナルのサンプル名(の略称)で対応関係を表すとsrp016842_samplename.txtのようになっていることがわかります。
尚このファイルはマッピング時の入力ファイルとしても用います。
Step2. カイコゲノム配列をダウンロード:
カイコゲノム配列はBSgenomeパッケージとして提供されていないため、自力で入手する必要があります。
手順としては、農業生物資源研究所(NIAS)が提供しているカイコゲノム配列のウェブページからIntegrated sequences (integretedseq.txt.gz)
をダウンロードし、解凍します。解凍後のファイル名は"integretedseq.txt"となりますが、拡張子を".txt"から".fa"に変更して、"integretedseq.fa"としたものを作業ディレクトリ(「デスクトップ」上の「SRP016842」フォルダ)上にコピーしておきます。
Step3. small RNA-seqデータの前処理:
原著論文(Nie et al., 2013)中では、アダプター配列やクオリティの低いリードを除去したのち、ゲノムにマッピングしたと書いてあります。
アダプター配列情報はどこにも書かれていませんでしたが、Table S2中のアダプター配列除去後の最も短いリードが18 nt (例:"GCAGTCGTGGCCGAGCGG")であり、
「この18 nt」と「この配列を含む生リード配列の差分」がアダプター配列ということになります。
詳細な情報は書かれていませんでしたが、おそらくアダプター配列は"TGGAATTCTCGGGTGCCAAGGAACTCCAGTC..."という感じだろうと推測できます。
ここでは、ダウンロードした"SRR609266.fastq.gz"ファイルを入力として、
1塩基ミスマッチまで許容して(推定)アダプター配列除去を行ったのち、"ACGT"のみからなる配列(許容するN数が0)で、
配列長が18nt以上のものをフィルタリングして出力しています。
in_f <- "SRR609266.fastq.gz"
out_f <- "SRR609266_p.fastq.gz"
param_adapter <- "TGGAATTCTCGGGTGCCAAGGAACTCCAGTC"
param_mismatch <- 1
param_nBases <- 0
param_minLength <- 18
library(QuasR)
res <- preprocessReads(filename=in_f,
outputFilename=out_f,
Rpattern=param_adapter,
max.Rmismatch=rep(param_mismatch, nchar(param_adapter)),
nBases=param_nBases,
minLength=param_minLength)
res
前処理実行後のresオブジェクトを眺めると、入力ファイルのリード数が11928428であり、
アダプター配列除去後に18nt未満の長さになってしまったためにフィルタリングされたリード数が157229、
"N"を1つ以上含むためにフィルタリングされたリード数が21422あったことがわかります。ここでは配列長分布は得ておりませんが、
出力ファイルを展開して配列長分布を調べると原著論文中のTable S1と似た結果になっていることから、ここでの処理が妥当であることがわかります。
Step4. カイコゲノムへのマッピングおよびカウントデータ取得:
マップしたい前処理後のFASTQファイル名("SRR609266_p.fastq.gz")およびその任意のサンプル名を記述したsrp016842_samplename.txtを作業ディレクトリに保存したうえで、下記を実行します。
Step2でダウンロードしたintegretedseq.faへマッピングしています。
basic alignerの一つであるBowtieを内部的に用いており、
ここではマッピング時のオプションを"-m 1 -v 0"とし、「ミスマッチを許容せず1ヶ所にのみマップされるもの」をレポートしています。
ミスマッチを許容していないため、--best --strataというオプションは事実上意味をなさないためにつけていません。
QuasRのマニュアル中のようにalignmentParameterオプションは特に指定せず(デフォルト)、50ヶ所にマップされるリードまでをレポートする
"maxHits=50"オプションをつけるという思想もあります。
マシンパワーにもよりますが、20分程度で終わると思います。
マップ後 | カウント情報取得 | single-end | ゲノム | アノテーション有 | QuasR(Gaidatzis_2015)の記述内容と基本的に同じです。
in_f1 <- "srp016842_samplename.txt"
in_f2 <- "integretedseq.fa"
out_f1 <- "srp016842_QC.pdf"
out_f2 <- "srp016842_other_info1.txt"
param_mapping <- "-m 1 -v 0"
library(QuasR)
library(GenomicAlignments)
library(Rsamtools)
time_s <- proc.time()
out <- qAlign(in_f1, in_f2,
alignmentParameter=param_mapping)
time_e <- proc.time()
qQCReport(out, pdfFilename=out_f1)
tmpfname <- out@alignments[,1]
tmpsname <- out@alignments[,2]
for(i in 1:length(tmpfname)){
k <- readGAlignments(tmpfname[i])
m <- reduce(granges(k))
tmpcount <- summarizeOverlaps(m, tmpfname[i])
count <- assays(tmpcount)$counts
colnames(count) <- tmpsname[i]
tmp <- cbind(as.data.frame(m), count)
out_f <- sub(".bam", "_range.txt", tmpfname[i])
write.table(tmp, out_f, sep="\t", append=F, quote=F, row.names=F, col.names=T)
out_f <- sub(".bam", "_range.fa", tmpfname[i])
fasta <- getSeq(FaFile(in_f2), m)
h <- as.data.frame(m)
names(fasta) <- paste(h[,1],h[,2],h[,3],h[,4],h[,5], sep="_")
writeXStringSet(fasta, file=out_f, format="fasta", width=50)
}
sink(out_f2)
cat("1. Computation time for mapping (in second).\n")
time_e - time_s
cat("\n\n2. Options used for mapping.\n")
out@alignmentParameter
cat("\n\n3. Alignment statistics.\n")
alignmentStats(out)
cat("\n\n4. Session info.\n")
sessionInfo()
sink()
無事マッピングが終了すると、指定した2つのファイルが生成されているはずです。
- QCレポートファイル(srp016842_QC.pdf):Quality Controlレポートです。よく利用されるFastQCのようなものです。
- その他の各種情報ファイル(srp016842_other_info1.txt):論文作成時に必要な、マッピング時に用いたオプション情報、マップされたリード数、Rおよび用いたパッケージのバージョン情報などを含みます。
この他にも以下に示すような形式のファイルがサンプルごとに自動生成されます。以下に示すファイル名中の"fa03ced5b37"に相当する部分はランダムな文字列からなり、サンプルごと、そして実行するたびに異なります。
理由は、同じ入力ファイルを異なるパラメータやリファレンス配列にマッピングしたときに、間違って上書きしてしまうのを防ぐためです。
- SRP016842:Nie et al., BMC Genomics, 2013
- SRAdb:Zhu et al., BMC Bioinformatics, 2013
- QuasR:Gaidatzis et al., Bioinformatics, 2015
- Bowtie:Langmead et al., Genome Biol., 2009
- GenomicAlignments:Lawrence et al., PLoS Comput. Biol., 2013
- Biostrings:原著論文なし
- Rsamtools
リンク集
- R
- Bioconductor:Gentleman et al., Genome Biol., 2004
- CRAN
- RjpWiki
- R Tips(竹澤様)
- BioEdit(フリーの配列編集ソフト)
- BioMart:Smedley et al., BMC Genomics, 2009
- DDBJ Read Annotation Pipeline:Nagasaki et al., DNA Res., 2013
- BioStar:Parnell et al., PLoS Comput Biol., 2011
- SEQanswers:Li et al., Bioinformatics, 2012
- NGS WikiBook:Li et al., Brief Bioinform., 2013
- HT Sequence Analysis with R and Bioconductor
- RNA-Seq Blog
- RNA-Seq Tutrial:Griffith et al., PLoS Comput Biol., 2015
SRA系
ソフト系
自分用
- オブジェクトの全消去は、rm(list = ls())
- 「"...\Rgui.exe" LANGUAGE=en」で英語環境にできる
- installed.packages()でパッケージがインストールされている場所を知ることができる。
Winは、「ライブラリ/ドキュメント/R/win-library/3.1/」などに存在する。
Macは、「/Library/Frameworks/R.framework/Versions/3.1/Resources/library」などに存在する。
- Macで「逆スラッシュ」を出したい場合は、「Alt + \」。
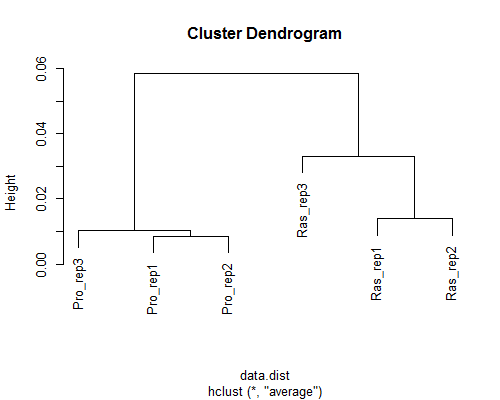{kind=link}
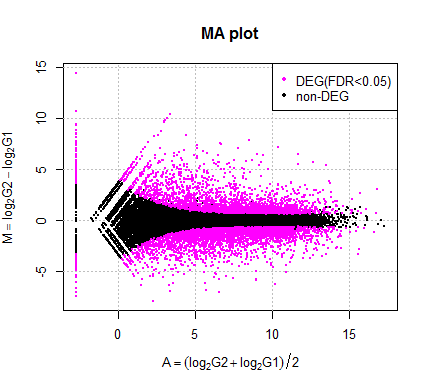{kind=link}
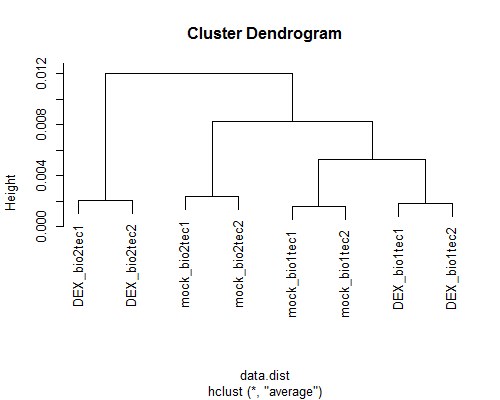{kind=link}
{kind=link}