第20回のウェブ資料
最終更新: 2023/04/30
はじめに(W1)
- 乳酸菌NGS連載第19回のPDF
- 乳酸菌NGS連載第18回のPDF
- JSLAB20.Rmd
- RStudio(2022年にPositに改名)
W1.1:Rパッケージのロード
このhtmlの作成に用いた元ファイルはJSLAB20.Rmdです。ただのテキストファイルですので、テキストエディタやRStudio(Positに改名)で開くことができます。JSLAB20.Rmdを開くと、第19回の本文中で解説した以外のチャンクオプションを使用していることがわかります。例えば、第19回のW05で解説したinclude=FALSEオプションは「チャンクをまるまるレポートに含めない」である一方、今回用いているmessage=FALSEオプションは「パッケージのロード時に表示されるメッセージをレポートに含めない」だけです。したがって、以下で示されているように、どのパッケージを使ったのかがレポートに含まれることになります。今回もこのような小技(Tips)を含めていきます。
library(TCC)
library(MBCluster.Seq)
library(openxlsx)
library(ggplot2)
library(ggsci)
library(reshape2)W1.2:ファイルの読込
入力ファイル(JSLAB18.xlsx)は、連載第18回から用いているLactobacillus
rhamnosus GG (LGG)のRNA-seqカウントデータ(Bang et al., J
Microbiol Biotechnol., 2018)です。連載第15回のW12-1のxlsxファイル(Kallisto_9samples.xlsx)を、読み込み時のオプションを減らすべく、データ以外の部分を削ったものです。連載第18回の図5でも示されていますが、2,949遺伝子×9サンプルの数値行列データです。以下では、まずread.xlsx関数を用いて読み込んだ入力ファイルの情報をdataオブジェクトに格納し、dim関数を用いてdataオブジェクトの行数と列数を表示させています。なお、読み込み時にオプションとしてrowNames=TRUEをつけているので、入力ファイル中の最初の1列目の情報が行名として取り扱われます。また、ここには明示されていませんがread.xlsx関数のデフォルトはcolNames=TRUEですので、最初の1行目が列名として取り扱われます。
data <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
dim(data)## [1] 2949 9W1.3:実験デザインと群ラベル情報の確認
連載第18回のp97でも概要が示されていますが、このデータはLactobacillus rhamnosus GG (LGG)の酸ストレス応答を調べたカウントデータです。以下に示す計3状態のRNA-seqデータを、各状態につき3反復ずつ取得して比較しています(Bang et al., J Microbiol Biotechnol., 2018)。
colnames(data)## [1] "pH4.5_1h_1" "pH4.5_1h_2" "pH4.5_1h_3" "pH4.5_24h_1" "pH4.5_24h_2"
## [6] "pH4.5_24h_3" "pH7_CCG_1" "pH7_CCG_2" "pH7_CCG_3"生物学的な反復と技術的な反復
- Zakharkin et
al., BMC Bioinformatics, 2005
Abstractの一番最後の文章(Correlations between technical replicates were consistently higher than between biological replicates.)からも技術的な反復データのほうが生物学的な反復データよりもデータの類似度が高いことがわかります。
- Bang et al., J Microbiol Biotechnol., 2018
解析データの記号表現(W2)
- Archer et al.,
BMC Bioinformatics, 2004
Fig. 1を見ると、横軸がMean、縦軸がVarianceになっていることがわかります。 - Anders and
Huber, Genome Biol., 2010
Fig. 1aがMean-Variance plotです。 - 門田幸二
著 (金明哲 編), シリーズUseful R ⑦ トランスクリプトーム解析,
2014
p121~129の3.2.4項ではマイクロアレイデータの性質が、そしてp145~165の3.3.4項ではRNA-seqカウントデータの性質がそれぞれ解説されています。本稿で用いている乳酸菌RNA-seqカウントデータの性質(平均-分散プロット結果)が「負の二項分布(Negative Binomial model; NB model)」に従うということが、この書籍のp163に見えている図3-38との類似性(比較対象はp159の図3-35)からもわかると思います。原稿中の「サンプル(反復または列)間の総カウント数の違いによる影響を排除する必要」性や具体的なコードについては、p158でも言及しています。
W2.1:列ごとのカウントの総和
2,949行×9列からなるdataオブジェクトを入力として、列ごとの総和(column
Sum)を算出するcolSums関数を実行しています。全部で9列の行列データなので、出力は9個の要素からなるベクトルになります。計9個しかないので比較的短時間で、最小がpH4.5_1h_1の233195、最大がpH4.5_24h_2の2641162であることがわかります。
colSums(data)## pH4.5_1h_1 pH4.5_1h_2 pH4.5_1h_3 pH4.5_24h_1 pH4.5_24h_2 pH4.5_24h_3
## 233195 840602 1072851 938788 2641162 1176816
## pH7_CCG_1 pH7_CCG_2 pH7_CCG_3
## 2381981 1758968 569947W2.2:Tips(max, min, range関数)
以下の最初の3つは、colSums関数実行結果であるcolSums(data)を入力として、その最大値、最小値、その両方(正確には範囲)を返すmax,
min,
range関数を実行しています。9つの要素からなる数値ベクトル程度であれば、もちろんみればわかります。しかし全体像がぱっとわかる程度もので、このような基本関数を使いこなせるようにしておくとよいと思います。なお、最後は最大/最小を算出し、10倍以上の違いがあることを示しています。
max(colSums(data))## [1] 2641162min(colSums(data))## [1] 233195range(colSums(data))## [1] 233195 2641162max(colSums(data))/min(colSums(data))## [1] 11.32598W2.3:Tips(order関数とオプション)
数値ベクトルを入力としてorder関数を実行すると、(デフォルトはdecreasing=Fなので)要素番号を値の昇順(小
\(\rightarrow\)
大)に並べ替えた結果が返されます。一番左側に見えている1は、入力ベクトルであるcolSums(data)中の1番目の要素が最小値(つまり233195)を持つことを意味します。同様に、一番右側に見えている5は、入力ベクトルであるcolSums(data)中の5番目の要素が最大値(つまり2641162)を持つことを意味します。なお、order関数実行時にdecreasing=Tをつけて実行すると、降順(大
\(\rightarrow\)
小)に並べ替えた結果が返されます。なお、要素番号はインデックス(index)ともいいます。
order(colSums(data))## [1] 1 9 2 4 3 6 8 7 5order(colSums(data), decreasing=T)## [1] 5 7 8 6 3 4 2 9 1- 乳酸菌NGS連載第18回のPDF
- 乳酸菌NGS連載第19回のPDF
- Osabe et al.,
BMC Bioinformatics, 2021
2ページ目の一番最後の行で、「… \(k\)th column as that in the non-DEG cluster, where \(k\) = argmin(||\(\mu_1\)||\(_2\), …, ||\(\mu_K\)||\(_2\)).」のように記載していることがわかります。
W2.4:Tips(which.max, which.min関数)
いわゆるargmaxに相当するのがwhich.max、argminに相当するのがwhich.minです。デフォルトオプションでorder関数を実行した結果の一番右側に見えている5を得ることを目的とした関数がwhich.max、一番左側に見えている1を得ることを目的とした関数がwhich.minだという理解でもよいです。
which.max(colSums(data))## pH4.5_24h_2
## 5which.min(colSums(data))## pH4.5_1h_1
## 1W2.5:本文と同じ記号に揃える
行列をA、遺伝子数をG,
サンプル数をS、遺伝子名をg、サンプル名をsとするコマンド群(スクリプト)です。行列の行数を返す関数がnrow、列数を返すのがncolです。また、行列の行名を返すのがrownames、列名を返すのがcolnamesです。なお、rownamesが有効なのは、read.xlsx関数で入力ファイルを読み込む際にオプションとしてrowNames=TRUEをつけているからです。また、colnamesが有効なのは、read.xlsx関数のデフォルトはcolNames=TRUEであり、入力ファイル中の最初の1行目が列名として取り扱われるようにしているからです。なお、head関数は、ベクトルを入力として与えると、(デフォルトはn=6なので)最初の6個分の要素が表示されます。
A <- data
G <- nrow(A)
S <- ncol(A)
g <- rownames(A)
s <- colnames(A)
G## [1] 2949S## [1] 9head(g)## [1] "EBG00001128470" "EBG00001128476" "EBG00001128500" "EBG00001128509"
## [5] "EBG00001128529" "LGG_00001"head(s)## [1] "pH4.5_1h_1" "pH4.5_1h_2" "pH4.5_1h_3" "pH4.5_24h_1" "pH4.5_24h_2"
## [6] "pH4.5_24h_3"総カウント数の計算(W3)
W3.1:総カウント数を算出する数式
式(1)
\(j\)列目のサンプル\(s_j\)の総カウント数\(T_j\)を算出する式です。
\[ T_j = \sum_{i=1}^G a_{i, j} \tag{1} \]\(1\)列目の総カウント数の算出例
\(1\)列目のサンプル\(s_1\)(つまりpH4.5_1h_1)の総カウント数\(T_1\)を算出する具体例です。
\[ \begin{aligned} T_1 &= \sum_{i=1}^{2949} a_{i, 1} \\ &= a_{1, 1} + a_{2, 1} + \ldots + a_{2948, 1} + a_{2949, 1} \\ &= 21 + 8 + \ldots + 121 + 1585 \\ &= 233195 \\ \end{aligned} \]\(5\)列目の総カウント数の算出例
\(5\)列目のサンプル\(s_5\)(つまりpH4.5_24h_2)の総カウント数\(T_5\)を算出する具体例です。
\[ \begin{aligned} T_5 &= \sum_{i=1}^{2949} a_{i, 5} \\ &= a_{1, 5} + a_{2, 5} + \ldots + a_{2948, 5} + a_{2949, 5} \\ &= 323 + 318 + \ldots + 1846 + 10940 \\ &= 2641162 \\ \end{aligned} \]\(9\)列目の総カウント数の算出例
\(9\)列目のサンプル\(s_9\)(つまりpH7_CCG_3)の総カウント数\(T_9\)を算出する具体例です。
\[ \begin{aligned} T_9 &= \sum_{i=1}^{2949} a_{i, 9} \\ &= a_{1, 9} + a_{2, 9} + \ldots + a_{2948, 9} + a_{2949, 9} \\ &= 124 + 27 + \ldots + 672 + 6157 \\ &= 569947 \\ \end{aligned} \]
W3.2:Tips(length, mean関数)
colSums(data)実行結果をTというオブジェクトに格納し、ベクトルTの要素数を返すlength関数や、平均値を返すmean関数を実行しています。この場合のベクトルの要素数である9は、2,949遺伝子×9サンプルの数値行列であるdataオブジェクトの列数に相当します。平均カウント数は約130万であることがわかります。
T <- colSums(data)
length(T)## [1] 9mean(T)## [1] 1290479W3.3:\(u\) = argmax()のあたり
W2.4で述べたように、Rではargmaxに相当する関数はwhich.maxですので、それを利用してuの値を得たのち、サンプル名情報を格納したsオブジェクトのインデックスとしてuを利用しています。
u <- which.max(T)
u## pH4.5_24h_2
## 5s[u]## [1] "pH4.5_24h_2"CPMの計算(W4)
W4.1:CPMを算出する数式
- 式(2)
\(i\)行\(j\)列目のCPM正規化後のカウント値\(a_{i, j}^{norm}\)を、正規化前のカウント値\(a_{i, j}\)と\(j\)列目のサンプル\(s_j\)の総カウント数\(T_j\)から算出する式です。
\[ a_{i, j}^{norm} = a_{i, j} \times \frac{1000000}{T_j} \tag{2} \] - \(1\)行\(1\)列目のCPM正規化後のカウント値\(a_{1, 1}^{norm}\)の算出例
\(1\)列目なので、\(1\)列目の総カウント数\(T_1\) = 233195を利用しています。
\[ \begin{aligned} a_{1, 1}^{norm} &= a_{1, 1} \times \frac{1000000}{T_1} \\ &= 21 \times \frac{1000000}{233195} \\ &= 90.053 \\ \end{aligned} \] - \(2\)行\(1\)列目のCPM正規化後のカウント値\(a_{2, 1}^{norm}\)の算出例
\(1\)列目なので、\(1\)列目の総カウント数\(T_1\) = 233195を利用しています。
\[ \begin{aligned} a_{2, 1}^{norm} &= a_{2, 1} \times \frac{1000000}{T_1} \\ &= 8 \times \frac{1000000}{233195} \\ &= 34.306 \\ \end{aligned} \] - \(2947\)行\(2\)列目のCPM正規化後のカウント値\(a_{2947, 2}^{norm}\)の算出例
\(2\)列目なので、\(2\)列目の総カウント数\(T_2\) = 840602を利用しています。
\[ \begin{aligned} a_{2947, 2}^{norm} &= a_{2947, 2} \times \frac{1000000}{T_2} \\ &= 212 \times \frac{1000000}{840602} \\ &= 252.200 \\ \end{aligned} \] - \(2949\)行\(2\)列目のCPM正規化後のカウント値\(a_{2949, 2}^{norm}\)の算出例
\(2\)列目なので、\(2\)列目の総カウント数\(T_2\) = 840602を利用しています。
\[ \begin{aligned} a_{2949, 2}^{norm} &= a_{2949, 2} \times \frac{1000000}{T_2} \\ &= 6737 \times \frac{1000000}{840602} \\ &= 8014.494 \\ \end{aligned} \]
W4.2:正規化係数部分の説明
式(2)の中で、正規化係数に相当するのは\(\frac{1000000}{T_j}\)です。これは数値ベクトルであり、要素数は9個であることがわかります。総カウント数情報を含むベクトル\(T\)(オブジェクト名は同じT)の平均は1290479、行列\(A\)(オブジェクト名は同じA)の行数と列数は2949と9、colSums(A)は\(T\)と同じ内容になります。
1000000/T## pH4.5_1h_1 pH4.5_1h_2 pH4.5_1h_3 pH4.5_24h_1 pH4.5_24h_2 pH4.5_24h_3
## 4.2882566 1.1896236 0.9320959 1.0652032 0.3786212 0.8497505
## pH7_CCG_1 pH7_CCG_2 pH7_CCG_3
## 0.4198186 0.5685152 1.7545491mean(T)## [1] 1290479dim(A)## [1] 2949 9colSums(A)## pH4.5_1h_1 pH4.5_1h_2 pH4.5_1h_3 pH4.5_24h_1 pH4.5_24h_2 pH4.5_24h_3
## 233195 840602 1072851 938788 2641162 1176816
## pH7_CCG_1 pH7_CCG_2 pH7_CCG_3
## 2381981 1758968 569947
以下は、正規化の実行と正規化後のデータの確認部分です。sweep関数の部分が本文中の式(2)に相当します。sweep関数部分は、入力である行列\(A\)の(x=A)、各列に対して(MARGIN=2)、正規化係数である1000000/Tを(STATS=1000000/T)、掛けよ(FUN="*")という命令を行った結果を行列\(A^{norm}\)に格納せよ(Rでは右上に添え字をつけられないのでAnormというオブジェクト名にしています)、ということをやっています。その後、CPM正規化によって得られたAnormオブジェクトの行数と列数が元のAと同じであることを示し、その総カウント数が全ての列で1000000(つまり1e+06)であるところまでを示しています。なお、式(2)自体は行列\(A\)の各要素\(a_{i,
j}\)の値を変換するものですが、Rでは行列そのものを入力として与えている点が異なります。
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
dim(Anorm)## [1] 2949 9colSums(Anorm)## pH4.5_1h_1 pH4.5_1h_2 pH4.5_1h_3 pH4.5_24h_1 pH4.5_24h_2 pH4.5_24h_3
## 1e+06 1e+06 1e+06 1e+06 1e+06 1e+06
## pH7_CCG_1 pH7_CCG_2 pH7_CCG_3
## 1e+06 1e+06 1e+06平均と分散を算出(W5)
W5.1 クラスラベル情報の作成
群ごとの反復数を整数ベクトルとして与えた結果をnkオブジェクトに格納しています。これが本文中の\(n_k\)に相当するものです。ここでは全て同じ数値なのでわかりずらいかもしれませんが、例えばc(10, 7, 4、6)と指定すると、群\(1\)の反復数\(n_1
= 10\)、群\(2\)の反復数\(n_2 = 7\)、群\(3\)の反復数\(n_3
= 4\)、そして群\(4\)の反復数\(n_4
= 6\)だと宣言していることになります。なお、群数\(K\)(Kオブジェクト)はlength(nk)で自動的に定めることができます。nkオブジェクトの要素数を返すlength関数の実行結果をKに代入(格納)しているだけです。このあたりは本文中の説明と実際の作業に若干解離がありますが、実装というのはそんなものです。Lを作成しているところが、クラスラベル\(L\)の作成部分になります。ちょっとややこしいので、とりあえずは「nkの部分さえちゃんと指定すれば、最終的にクラスラベル\(L\)(Lオブジェクト)を得られる」という理解でよいです。
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
L## [1] 1 1 1 2 2 2 3 3 3なお、上記のスクリプトはあくまでもnkオブジェクトさえ与えれば、あとは自動的にクラスラベルLを得られるようにするだけのものです。理解しやすさを優先すれば、以下のような感じでrep関数のみを用いてべた書きでも全く問題ありません。
L <- c(rep(1, 3), rep(2, 3), rep(3, 3))
L## [1] 1 1 1 2 2 2 3 3 3もう1つ、as.vectorとmapplyを使わずに、forループを用いるやり方も示しておきます。
nk <- c(3, 3, 3)
K <- length(nk)
L <- NULL
for(i in 1:K){
L <- c(L, rep(i, nk[i]))
}
L## [1] 1 1 1 2 2 2 3 3 3念のため、mapply関数について説明しておきます。これは2つ以上の変数を入力として何か関数を実行したいときに用います。この場合は、「rep関数を、xオプションとtimesオプションという2つの変数を与えて、うまくクラスラベル情報のベクトルを作成する」目的で利用しています。以下では反復数を変えて挙動をわかりやすくしています。mapply(rep, 1:K, nk)実行結果はリスト形式と呼ばれるものですので、これを入力としてas.vector関数を実行してあげれば、「ベクトルとして(as
vector)」結果が返されます。mapplyは、rep(1, times=4)とrep(2, times=3)とrep(3, times=5)
をそれぞれ実行し、それらの結果をリスト形式で返しているのだと理解すればよいです。
nk <- c(4, 3, 5)
K <- length(nk)
mapply(rep, 1:K, nk)## [[1]]
## [1] 1 1 1 1
##
## [[2]]
## [1] 2 2 2
##
## [[3]]
## [1] 3 3 3 3 3W5.2 群ごとにカウントの平均を計算
まずはR再起動後でもリカバリできるように、入力ファイルの読込からクラスラベル作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))以下は、群ごとの遺伝子の平均カウントを算出した結果を行列Mに格納していくスクリプトです。入力はCPM正規化後のカウントデータ行列Anormです。Anorm[, L==i]は、平均カウント数を計算したい特定の群のみからなる行列(つまりサブセット)の抽出に相当します。例えばiが2のときはクラスラベルLが2に相当する列のみからなる行列のサブセットを抽出していることになります。そしてそれを入力として、rowMeans関数を実行して行ごとの平均(rowのmean)を計算しています。rowMeansの実行結果自体は数値ベクトルですので、群の数だけの数値ベクトルが得られることになります。cbind関数は、これらの要素数が同じ数値ベクトルを列方向で結合(column
bind)するために利用しています。尚、forループの上にあるM <- NULLは、(これからMというオブジェクトに結果を格納していきたいので)Mというプレースホルダを作成しているだけです。Mという入れ物(それがプレースホルダという概念)を予め確保しているだけだという理解で構いません。
M <- NULL
for(i in 1:K){
M <- cbind(M, rowMeans(Anorm[, L==i]))
}
dim(M)## [1] 2949 3head(M)## [,1] [,2] [,3]
## EBG00001128470 219.35372 139.9754 163.20042
## EBG00001128476 88.02994 116.9911 45.61295
## EBG00001128500 26661.18736 23549.0411 35571.20466
## EBG00001128509 259.62436 166.7145 190.11327
## EBG00001128529 0.00000 0.0000 0.00000
## LGG_00001 419.03678 412.6478 318.10139以下は、上記スクリプトをrowMeansの代わりに、apply関数を用いるやり方です。applyは、行ごと(MARGIN=1)あるいは列ごと(MARGIN=2)に任意の関数を実行できますので、meanという一般的な平均を計算する関数を利用しています。上の結果と見比べると、確かに同じ結果が得られていることがわかります。なお、以下はapply関数実行時にオプション名を明記していますが、apply(Anorm[, L==i], 1, mean)のように省略してもかまいません。
M <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
}
dim(M)## [1] 2949 3head(M)## [,1] [,2] [,3]
## EBG00001128470 219.35372 139.9754 163.20042
## EBG00001128476 88.02994 116.9911 45.61295
## EBG00001128500 26661.18736 23549.0411 35571.20466
## EBG00001128509 259.62436 166.7145 190.11327
## EBG00001128529 0.00000 0.0000 0.00000
## LGG_00001 419.03678 412.6478 318.10139最後に、計算例の解説です。例えば上で見えている行列Mの2行3列目の45.61295という値は、入力である行列Anormの2行目(つまり\(g_2\) =
EBG00001128476)の群3(つまりpH7_CCG群)の平均を計算した結果です。
head(Anorm)## pH4.5_1h_1 pH4.5_1h_2 pH4.5_1h_3 pH4.5_24h_1 pH4.5_24h_2
## EBG00001128470 90.05339 311.6814 256.3264 152.3241 122.2947
## EBG00001128476 34.30605 114.2039 115.5799 102.2595 120.4016
## EBG00001128500 28212.44023 24920.2357 26850.8861 24275.9814 21790.4089
## EBG00001128509 68.61211 396.1447 314.1163 170.4325 152.9630
## EBG00001128529 0.00000 0.0000 0.0000 0.0000 0.0000
## LGG_00001 450.26694 390.1965 416.6469 382.4080 422.1627
## pH4.5_24h_3 pH7_CCG_1 pH7_CCG_2 pH7_CCG_3
## EBG00001128470 145.3073 90.68082 181.35634 217.56409
## EBG00001128476 128.3123 41.14223 48.32379 47.37283
## EBG00001128500 24580.7331 27418.77454 38480.51812 40814.32133
## EBG00001128509 176.7481 137.70051 229.11162 203.52770
## EBG00001128529 0.0000 0.00000 0.00000 0.00000
## LGG_00001 433.3728 327.45853 309.27226 317.57339Anorm[2, L==3]## pH7_CCG_1 pH7_CCG_2 pH7_CCG_3
## EBG00001128476 41.14223 48.32379 47.37283(41.14223 + 48.32379 + 47.37283)/3## [1] 45.61295W5.3 群ごとにカウントの分散を計算
まずはR再起動後でもリカバリできるように、入力ファイルの読込から群ごとにカウントの平均作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
M <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
}分散\(V\)についても、平均のときと同じノリで算出することができます。以下ではVという行列として(分散なのでmeanではなくvar関数に変更して)作成しています。
V <- NULL
for(i in 1:K){
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
}
dim(V)## [1] 2949 3head(V)## [,1] [,2] [,3]
## EBG00001128470 13304.9755 246.7638 4.272069e+03
## EBG00001128476 2165.1651 178.4106 1.521660e+01
## EBG00001128500 2736641.8214 2342808.8423 5.120825e+07
## EBG00001128509 29046.4243 151.8007 2.223958e+03
## EBG00001128529 0.0000 0.0000 0.000000e+00
## LGG_00001 906.3969 717.2525 8.289425e+01- \(v_{6,2}\)の計算例 \[ \begin{aligned} v_{6, 2} &= \frac{1}{n_2 - 1} \sum_{j \mid l_j = 2} \bigl(a_{i,j}^{norm} - m_{6, 2}\bigr)^2 \\ &= \frac{1}{3 - 1} \Bigl( \bigl(a_{6,4}^{norm} - m_{6, 2}\bigr)^2 + \bigl(a_{6,5}^{norm} - m_{6, 2}\bigr)^2 + \bigl(a_{6,6}^{norm} - m_{6, 2}\bigr)^2 \Bigr) \\ &= \frac{1}{2} \Bigl( \bigl(382.408 - 412.648 \bigr)^2 + \bigl(422.163 - 412.648 \bigr)^2 + \bigl(433.373 - 412.648 \bigr)^2 \Bigr) \\ &= \frac{1}{2} \Bigl( 914.445 + 90.533 + 429.526 \Bigr) \\ &= 717.252 \\ \end{aligned} \]
平均と分散の関係性(W6)
W6.1 平均と分散の群ごとの要約統計量
まずはR再起動後でもリカバリできるように、入力ファイルの読込から群ごとのカウントの平均と分散の作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
M <- NULL
V <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
}以下は、図3と同じ結果を得るスクリプトです。
summary(M)## V1 V2 V3
## Min. : 0.00 Min. : 0.0 Min. : 0.0
## 1st Qu.: 32.24 1st Qu.: 44.0 1st Qu.: 20.2
## Median : 102.55 Median : 103.7 Median : 79.5
## Mean : 339.10 Mean : 339.1 Mean : 339.1
## 3rd Qu.: 268.25 3rd Qu.: 236.8 3rd Qu.: 253.8
## Max. :28133.00 Max. :53225.3 Max. :35572.6summary(V)## V1 V2 V3
## Min. : 0 Min. : 0 Min. : 0
## 1st Qu.: 77 1st Qu.: 17 1st Qu.: 22
## Median : 594 Median : 69 Median : 222
## Mean : 118342 Mean : 72765 Mean : 85000
## 3rd Qu.: 4888 3rd Qu.: 284 3rd Qu.: 2122
## Max. :32212918 Max. :180749285 Max. :51208247W6.2 負の二項分布(NB分布)のパラメータ\(\phi\)を得る
平均\(M\)と分散\(V\)の最初の6行分を表示するスクリプトです。
head(M)## [,1] [,2] [,3]
## EBG00001128470 219.35372 139.9754 163.20042
## EBG00001128476 88.02994 116.9911 45.61295
## EBG00001128500 26661.18736 23549.0411 35571.20466
## EBG00001128509 259.62436 166.7145 190.11327
## EBG00001128529 0.00000 0.0000 0.00000
## LGG_00001 419.03678 412.6478 318.10139head(V)## [,1] [,2] [,3]
## EBG00001128470 13304.9755 246.7638 4.272069e+03
## EBG00001128476 2165.1651 178.4106 1.521660e+01
## EBG00001128500 2736641.8214 2342808.8423 5.120825e+07
## EBG00001128509 29046.4243 151.8007 2.223958e+03
## EBG00001128529 0.0000 0.0000 0.000000e+00
## LGG_00001 906.3969 717.2525 8.289425e+01任意の群\(k\)における\(i\)行目の遺伝子の分散\(v_{i,k}\)は、\(v_{i,k} = m_{i,k} + \phi_{i,k} \times
m_{i,k}^2\)と表すことができます。これ式変形するとを\(\phi_{i,k} = \frac{v_{i,k} -
m_{i,k}}{m_{i,k}^2}\)となるので、FUNオプションのところでfunction(x){var(x) - mean(x))/mean(x)^2}という関数を定義して実行しています。
Phi <- NULL
for(i in 1:K){
Phi <- cbind(Phi, apply(X=Anorm[, L==i], MARGIN=1, FUN=function(x){(var(x) - mean(x))/mean(x)^2}))
}
dim(Phi)## [1] 2949 3head(Phi)## [,1] [,2] [,3]
## EBG00001128470 0.271959587 0.0054503107 0.154269370
## EBG00001128476 0.268042701 0.0044874537 -0.014609831
## EBG00001128500 0.003812477 0.0041821822 0.040442778
## EBG00001128509 0.427073383 -0.0005365914 0.056272060
## EBG00001128529 NaN NaN NaN
## LGG_00001 0.002775528 0.0017888648 -0.002324445同じ結果を得る別のやり方です。ここでは、func_phiという名前の関数を新たに定義して実行しています。
func_phi <- function(x){
phi_value <- (var(x) - mean(x))/mean(x)^2
return(phi_value)
}
Phi <- NULL
for(i in 1:K){
Phi <- cbind(Phi, apply(X=Anorm[, L==i], MARGIN=1, FUN=func_phi))
}
dim(Phi)## [1] 2949 3head(Phi)## [,1] [,2] [,3]
## EBG00001128470 0.271959587 0.0054503107 0.154269370
## EBG00001128476 0.268042701 0.0044874537 -0.014609831
## EBG00001128500 0.003812477 0.0041821822 0.040442778
## EBG00001128509 0.427073383 -0.0005365914 0.056272060
## EBG00001128529 NaN NaN NaN
## LGG_00001 0.002775528 0.0017888648 -0.002324445同じ結果を得るさらに別のやり方です。上記までのやり方だと\(\phi_{6,k}\)がNaN(Not a
Numberという意味)になってしまいます。この原因は\(\frac{v_{i,k} -
m_{i,k}}{m_{i,k}^2}\)の分母が0であることに起因します。そこで、「もし\(m_{i,k} = 0\)なら\(\phi_{i,k} =
0\)とし、それ以外(実質的には\(m_{i,k}
> 0\))の場合は普通に\(\frac{v_{i,k}
-
m_{i,k}}{m_{i,k}^2}\)を計算せよ。」という条件分岐を行っています。こういう条件分岐などちょっとややこしい場合に、特に見やすさという点で関数を別に定義するメリットがあると思います。
func_phi <- function(x){
if(mean(x) == 0){ phi_value <- 0 }
else{ phi_value <- (var(x) - mean(x))/mean(x)^2 }
return(phi_value)
}
Phi <- NULL
for(i in 1:K){
Phi <- cbind(Phi, apply(X=Anorm[, L==i], MARGIN=1, FUN=func_phi))
}
dim(Phi)## [1] 2949 3head(Phi)## [,1] [,2] [,3]
## EBG00001128470 0.271959587 0.0054503107 0.154269370
## EBG00001128476 0.268042701 0.0044874537 -0.014609831
## EBG00001128500 0.003812477 0.0041821822 0.040442778
## EBG00001128509 0.427073383 -0.0005365914 0.056272060
## EBG00001128529 0.000000000 0.0000000000 0.000000000
## LGG_00001 0.002775528 0.0017888648 -0.002324445W6.3 NB分布に従うことを確認する
ここではまず、本文中の記載内容通りに\(i =
6, k =
2\)として平均M[i,k]とdispersionPhi[i,k]の値を表示させています。次に、set.seed関数を用いてタネ番号を1023に固定し、同じ乱数が発生するようにしています。その上で、rnbinom関数に必要な平均とdispersionの情報、そして乱数を発生させる数をn=8で指定しています。\(NB(m_{i,k},
\phi_{i,k})\)に従う乱数を発生させた結果をoutputオブジェクトに格納し、その中身の表示や分散をvar(output)として表示させています。V[i,k]は分散の期待値です。
i <- 6
k <- 2
M[i,k]## LGG_00001
## 412.6478Phi[i,k]## LGG_00001
## 0.001788865set.seed(1023)
output <- rnbinom(n=8, mu=M[i,k], size=1/Phi[i,k])
output## [1] 400 392 446 430 377 423 406 412var(output)## [1] 484.7857V[i,k]## LGG_00001
## 717.2525以下では、rnbinom関数をn=300,
72000,
そして172000で実行した結果として得られた乱数の分散をそれぞれ表示させています。そして最後にV[i,k]は分散の期待値を表示させています。この場合は、試行回数を増やすほど期待値に近い値が得られていることがわかります。
output <- rnbinom(n=300, mu=M[i,k], size=1/Phi[i,k])
var(output)## [1] 707.334output <- rnbinom(n=72000, mu=M[i,k], size=1/Phi[i,k])
var(output)## [1] 711.2591output <- rnbinom(n=172000, mu=M[i,k], size=1/Phi[i,k])
var(output)## [1] 716.4808V[i,k]## LGG_00001
## 717.2525描画:古典的な方法(W7)
ここでは、ggplot2を使わない昔ながらのプロットのやり方を示します。
W7.1 必要最小限のプロット
入力としては、ともに2949行×3列からなる平均Mと分散Vの同じ列を与えてプロットするのが基本形です。行列のsubsetting(サブセットを得ること)は、行列[行, 列]のような感じでコンマ(,)の左側で行の指定、右側で列の指定を行います。例えばk列目の情報を入力として与えたい場合はM[, k]とV[, k]とすればよいです。なお、実行結果で「4
個の正でない x
の値が対数プロットから除かれました」のような警告が出ていますが、これは「0のlogをとれない」というものです。それらはプロットされないだけですので気にしなくてかまいません。
k <- 1
plot(x=M[, k], y=V[, k], log="xy")## Warning in xy.coords(x, y, xlabel, ylabel, log): 4 個の正でない x
## の値が対数プロットから除かれました## Warning in xy.coords(x, y, xlabel, ylabel, log): 4 個の正でない y
## の値が対数プロットから除かれました
上記の警告メッセージは、たとえば平均M行列中のk列目において、要素(元)が0になっている数をsum関数で調べてみると、確かに4という結果が得られるので納得できると思います。
sum(M[,k] == 0)## [1] 4W7.2 gridの追加
以下のようにすればグリッドを追加できます。こうすることで、y=xの直線がどこらへんにあるかのイメージがつかめます。ちなみに(これは元ファイルであるJSLAB20.Rmdを見ないとわかりませんが)警告メッセージをこのhtmlファイルに出力させないオプション(warning=FALSE)をつけています。
k <- 1
plot(x=M[, k], y=V[, k], log="xy")
grid(col="gray", lty="dotted")
W7.3 ablineの追加
以下のようにすればy=xの直線をを追加できます。abline関数のオプションは、y=a+bxのa=0とb=1に対応します。
k <- 1
plot(x=M[, k], y=V[, k], log="xy")
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
W7.4 軸の範囲を指定
plot関数実行時に、x軸y軸の表示範囲を指定するxlimおよびylimオプションを追加しています。
k <- 1
axis_range <- c(1e-01, 1e+7)
plot(x=M[, k], y=V[, k], log="xy", xlim=axis_range, ylim=axis_range)
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
W7.5 点の形や大きさを指定
plot関数実行時に、プロットの形を指定するpchオプションと、大きさを指定するcexオプションを追加しています。pchは、数値と形が対応づいています。cexは、デフォルトの何倍かというのを指定するものです。例えば0.3なら通常の0.3倍の大きさにせよということです。
k <- 1
axis_range <- c(1e-01, 1e+7)
plot(x=M[, k], y=V[, k], log="xy", xlim=axis_range, ylim=axis_range, pch=20, cex=0.4)
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
W7.6 条件を満たす点を別の色に変更
さきほどまでのスクリプトの下に、まず「平均Mのほうが分散Vよりも大きい遺伝子をTRUE、それ以外をFALSEという条件判定」を行った結果をobjというオブジェクトに格納しています。このようなTRUE
or
FALSEのみから構成されるベクトルのことを論理値ベクトルといいます。次に、points関数を用いてobjがTRUEの遺伝子のみ、赤色(col="red")で点を追加しています。plot関数とpoints関数で用いているオプションがともに同じpch=20, cex=0.4からも予想できますが、ここで行っているのは、結局一度プロットしたもののうち、objがTRUEの要素のみ赤色に塗り替えているのだという理解でよいです。なお、プロットの下にsum(obj)の実行結果がありますが、これはTRUEとなった要素数が434個であったことを表示させているだけです。2949個中434個のみがy
= xの直線よりも右下にあるということですね。
k <- 1
axis_range <- c(1e-01, 1e+7)
plot(x=M[, k], y=V[, k], log="xy", xlim=axis_range, ylim=axis_range, pch=20, cex=0.4)
grid(col="gray", lty="dotted")
abline(a=0, b=1, col="gray")
obj <- (M[, k] > V[, k])
points(x=M[obj, k], y=V[obj, k], log="xy", xlim=axis_range, ylim=axis_range, pch=20, cex=0.4, col="red")
sum(obj)## [1] 434描画:ggplot2の基礎(W8)
ggplot2を用いて描画するやり方を示していきます。ggplot2は、全部で3群ある群ラベル(グループラベル)情報を含むデータを読み込んで群ごとに色分けできたりする機能が本来備わっています。しかしW8では、全部で3群(G1群、G2群、G3群)あるデータのうち、G1群(酸ストレス短期暴露群;pH4.5_1h群)に限定して、W7と似たようなノリで解説していきます。群ラベル情報を含む3群全てのデータを取り扱うのはW9です。R再起動後でもリカバリできるように、入力ファイルの読込から群ごとのカウントの平均と分散の作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
M <- NULL
V <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
}W8.1 入力データの準備
ggplot2は、入力としてデータフレーム形式のオブジェクトを用いるのが一般的であり、列名を指定してプロットに用いる情報を指定します。ここではまず、入力オブジェクトの「型」を返すclass関数を実行し、平均の情報を格納したMオブジェクト、および分散の情報を格納したVオブジェクトの「型」がいずれもデータフレーム(data.frame)形式ではなく「“matrix”
“array”」形式であることを認識します。次に、data.frame関数を用いて、2つの行列MとVのk列目の情報のみ、列名をつけて(それが”MEAN1=“や”VAR1=“をつけていることに相当します)マージした結果をdというオブジェクトにデータフレーム形式で格納しています。そして、再度dオブジェクトを入力としてclass関数を実行し、無事dオブジェクトの「型」がデータフレーム(data.frame)形式なっていることを確認しています。最後に、head関数でdオブジェクトの最初の3行分を表示させています。なお、列名をMEAN1やVAR1としているのは、今作成したこのdオブジェクトには、全部で3群(G1群、G2群、G3群)あるデータのうち、G1群(酸ストレス短期暴露群;pH4.5_1h群)に限定していることを強く意識させることを目的としているからです。
class(M)## [1] "matrix" "array"class(V)## [1] "matrix" "array"k <- 1
d <- data.frame(MEAN1=M[, k], VAR1=V[, k])
class(d)## [1] "data.frame"head(d, n=3)## MEAN1 VAR1
## EBG00001128470 219.35372 13304.976
## EBG00001128476 88.02994 2165.165
## EBG00001128500 26661.18736 2736641.821W8.2 必要最小限のプロット
W8.2.1 パッケージのロード
ggplot2は、より大きな枠組みであるtidyverseというパッケージの一部です。したがってtidyverseをインストール済みであれば、library(ggplot2)の代わりにlibrary(tidyverse)でも構いません。インストールから行いたい場合は、BiocManager::install("tidyverse", update=F)またはinstall.packages("tidyverse")で可能なはずです。もし「Error
in install.packages : Updating loaded
packages」のようなエラーメッセージがでてインストールできない場合は、おそらくtidyverse支配下のパッケージが既にロードされているためですので(たとえばlibrary(ggplot2)を実行済みの状況ということ)、たとえば起動中のRStudio(Positに改名)自体を物理的に再起動してやればうまくいきます。もしそれでもエラーが出てうまくいかない場合は、おそらくR本体のバージョンがちょっと古いことに起因する問題に遭遇していると予想されますので、CRANから最新版のRをインストールするところからやるとよいです。
library(ggplot2)W8.2.2 書き方1
まずはdオブジェクト中の、群1の平均(MEAN1列)をx軸に、そして群1の分散(VAR1列)をy軸として、散布図を描きます。
g <- ggplot(d, aes(x=MEAN1,y=VAR1))
g <- g + geom_point()
plot(g)
W8.2.3 書き方2
上と基本的に同じですが、plot関数の代わりにprint関数でも同じ結果が得られますよというだけです。
g <- ggplot(d, aes(x=MEAN1,y=VAR1))
g <- g + geom_point()
print(g)
W8.2.4 書き方3
上と基本的に同じですが、aes(x=MEAN1,y=VAR1)をggplot関数内でなくgeom_point関数内に配置しても構わないという例です。
g <- ggplot(d)
g <- g + geom_point(aes(x=MEAN1,y=VAR1))
print(g)
W8.2.5 書き方4
ヒトによっては「g <- g + geom_point(aes(x=MEAN1,y=VAR1))」という記載が非常に難解だという感想をもつかもしれません。しかしこれってよく見ると、geom_point(aes(x=MEAN1,y=VAR1))の内容をgに追加しているだけだということに気づきます。そう考えると以下のような書き方でも大丈夫だということがわかると思います。そして、このような書き方でもよいことに納得できれば、さきほどの「書き方3」でaes(x=MEAN1,y=VAR1)を記載する位置が重要でないことにも合点がいくと思います。
g <- ggplot(d) + geom_point(aes(x=MEAN1,y=VAR1))
print(g)
W8.2.6 書き方5
「書き方4」のようにすると、横長になってしまって見づらいヒトも出てくると思います。それを避けるべく、以下のように「行の最後を+で終える」書き方もあります。これもググるとわりと見かける書き方ですが、単に「+のあとで改行を入れているだけ」だということがよくわかると思います。なお、一番最後をprintからplotに変えていますが、特に深い意味はありません。ggplotはこんな感じでいろんなコマンドを足していくのが基本です。
g <- ggplot(d) +
geom_point(aes(x=MEAN1,y=VAR1))
plot(g)
W8.3 対数変換
W8.3.1 log10変換(やり方1)
列名に相当するMEAN1やVAR1のところに直接log10を書いているので違和感があるかもしれませんが、こうすることで元のdを変えずにlog10変換したものをプロットすることができます。
g <- ggplot(d) +
geom_point(aes(x=log10(MEAN1),y=log10(VAR1)))
plot(g)
W8.3.2 log10変換(やり方2)
以下のように、scale_x_log10()でx軸のスケールを、そしてscale_y_log10()でy軸のスケールをそれぞれlog10変換することができます。「Warning: Transformation introduced infinite values in continuous...」という警告メッセージが出ていることがわかります。これは、前述のように0の対数がとれずに無限大(infinite
values)になってしまうというものですので、それらがプロットされないという事実のみ正しく認識していればそれでOKです。
g <- ggplot(d) +
geom_point(aes(x=MEAN1,y=VAR1)) +
scale_x_log10() +
scale_y_log10()
plot(g)## Warning: Transformation introduced infinite values in continuous x-axis## Warning: Transformation introduced infinite values in continuous y-axis
W8.3.3 log10変換(やり方3)
(これは元ファイルであるJSLAB20.Rmdを見ないとわかりませんが)さきほどの「やり方2」で出現していた警告メッセージををこのhtmlファイルに出力させないオプション(warning=FALSE)をつけています。ついでに、後ほどgeom_point内でオプションを追加したいので、aes(x=MEAN1,y=VAR1)の位置を変更しています。
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point() +
scale_x_log10() +
scale_y_log10()
plot(g)
W8.4 背景のテーマを変更
W8.4.1 デフォルト(theme_gray)
デフォルトの背景テーマを明示しただけです。theme_gray()が追加されていることがわかりますが、デフォルトはこれです。
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_gray()
plot(g)
W8.4.2 白でグリッドありで外枠あり(theme_bw)
背景が白でグリッドありで外枠ありの場合はtheme_bw()にすればよいです。
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw()
plot(g)
W8.4.3 白でグリッドなしで外枠半分あり(theme_classic)
背景が白でグリッドなしで外枠半分ありの場合はtheme_classic()にすればよいです。
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_classic()
plot(g)
W8.4.4 白でグリッドありで外枠なし(theme_minimal)
背景が白でグリッドありで外枠なしの場合はtheme_minimal()にすればよいです。
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_minimal()
plot(g)
W8.5 ablineの追加
W7.3と同じく、y=xの直線を引きます。geom_ablineのintercept=0オプションが切片の値が0ということに、そしてslope=1が傾きが1であることの指定に相当します。なお、背景のテーマはtheme_bw()にしています。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue")
plot(g)
W8.6 軸の範囲を指定
W7.4と似た指定方法にしています。ggplotでは、coord_cartesianの中のオプションとして与えます。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range)
plot(g)
W8.7 点の形や大きさを指定
W7.5と似た部分です。これは既出のgeom_pointの中でshapeやsizeというオプションで与えます。もちろんcolorオプションで色も変えられます。
W8.7.1 変更例1(shape=11, size=1.5)
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=11, size=1.5, color="red") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range)
plot(g)
W8.7.2 変更例2(shape=8, size=1.2)
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=8, size=1.2, color="pink") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range)
plot(g)
W8.7.3 変更例3(shape=3, size=0.7)
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=3, size=0.7, color="lightgreen") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range)
plot(g)
W8.7.4 変更例4(shape=20, size=0.6)
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="lightblue") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range)
plot(g)
W8.8 軸ラベルの変更
W8.8.1 変更例1(labsで一気に変更)
labsを用いて、x軸とy軸のラベル、そしてタイトルを一気に変更する例です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)", title="hogege")
plot(g)
W8.8.2 変更例2(labsでx軸のみ変更)
labsを用いて、x軸のラベルのみ変更する例です。明示していない部分はデフォルト(たとえばy軸はVAR1)になります。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)")
plot(g)
W8.8.3 変更例3(ylabでy軸のみ変更)
ylabを用いてy軸のラベルのみ変更する例です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
ylab("Variance(Group1)")
plot(g)
W8.8.4 変更例4(ggtitleでタイトルを独立に変更)
ggtitleを用いてタイトルを変更する例です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
ylab("Variance(Group1)") +
ggtitle("nyuusannkin")
plot(g)
W8.9 図のサイズを変更
W8.9.1 R Markdown側で指定
これは、このhtmlの作成に用いた元ファイル(JSLAB20.Rmd)の対応するチャンクをみないとわかりませんが、fig.width=5, fig.height=5を追加しています。たしかに下で見えている画像をコピーしてパワポなどに貼り付けて眺めると、横幅、縦幅ともに同じ長さになっていることがわかります。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)")
plot(g)
W8.9.2 ggsaveでpngファイルに保存1
これは、一番最後のplot(g)の代わりにggsaveを用いてファイルに保存するやり方です。縦横のサイズは、単位をピクセルにして(units="px")、横幅が1500(width=1500)、縦幅が1400(height=1400)にしたうえで、png形式で(device="png")、JSLAB20.w8.9.2.pngというファイル名で保存しています。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)")
ggsave("JSLAB20.w8.9.2.png", g, units="px", width=1500, height=1400, device="png")得られたJSLAB20.w8.9.2.pngは、以下に見えているものと同じになります。

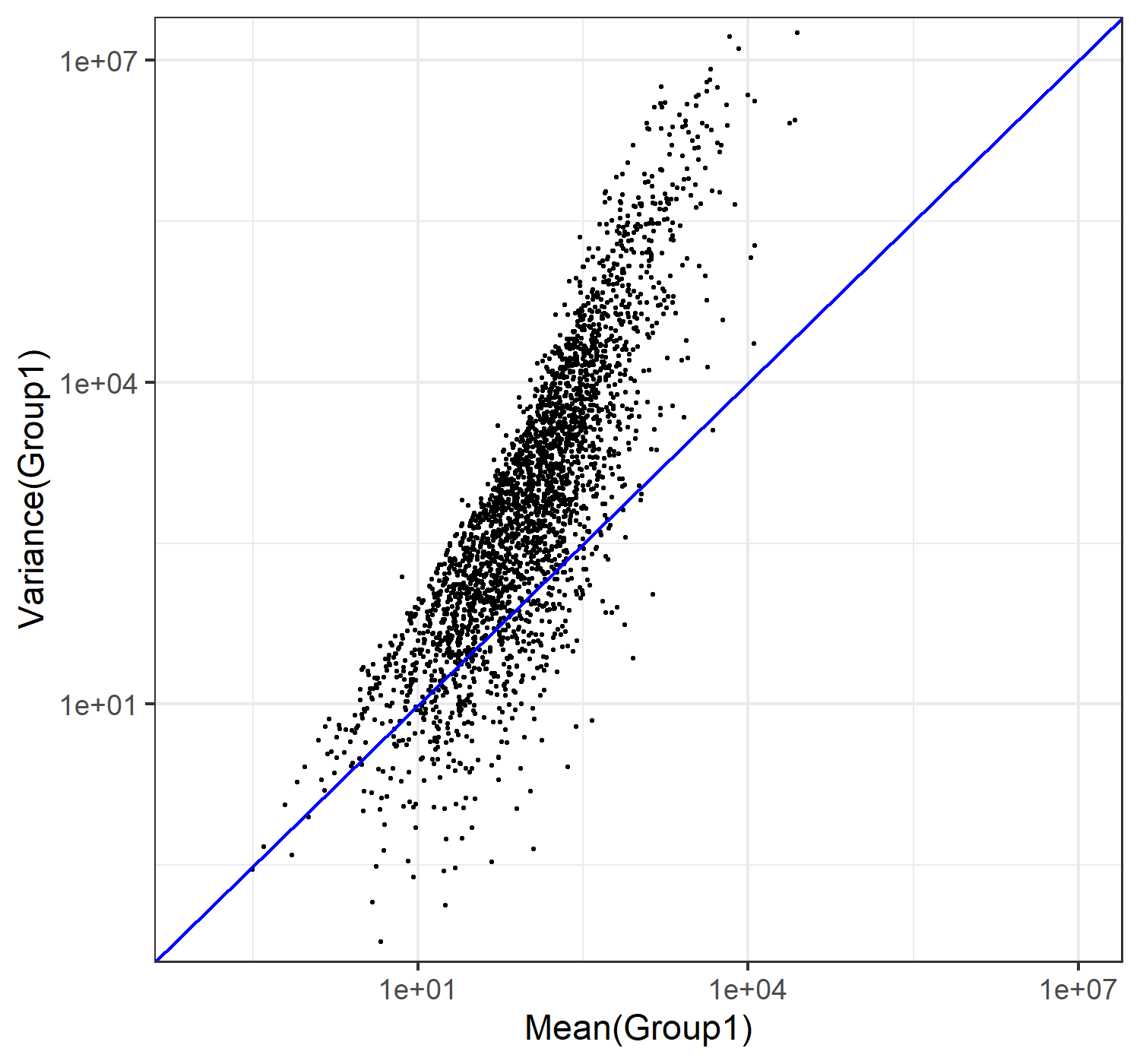{kind=link}
W8.9.3 ggsaveでpngファイルに保存2
さきほどの例と似ていますが、出力のところのオプションとしてunitsを明示せずにデフォルトのインチ(units="in"に相当)でやっています。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)")
ggsave("JSLAB20.w8.9.3.png", g, width=6, height=5, device="png")得られたJSLAB20.w8.9.3.pngは、以下に見えているものと同じになります。

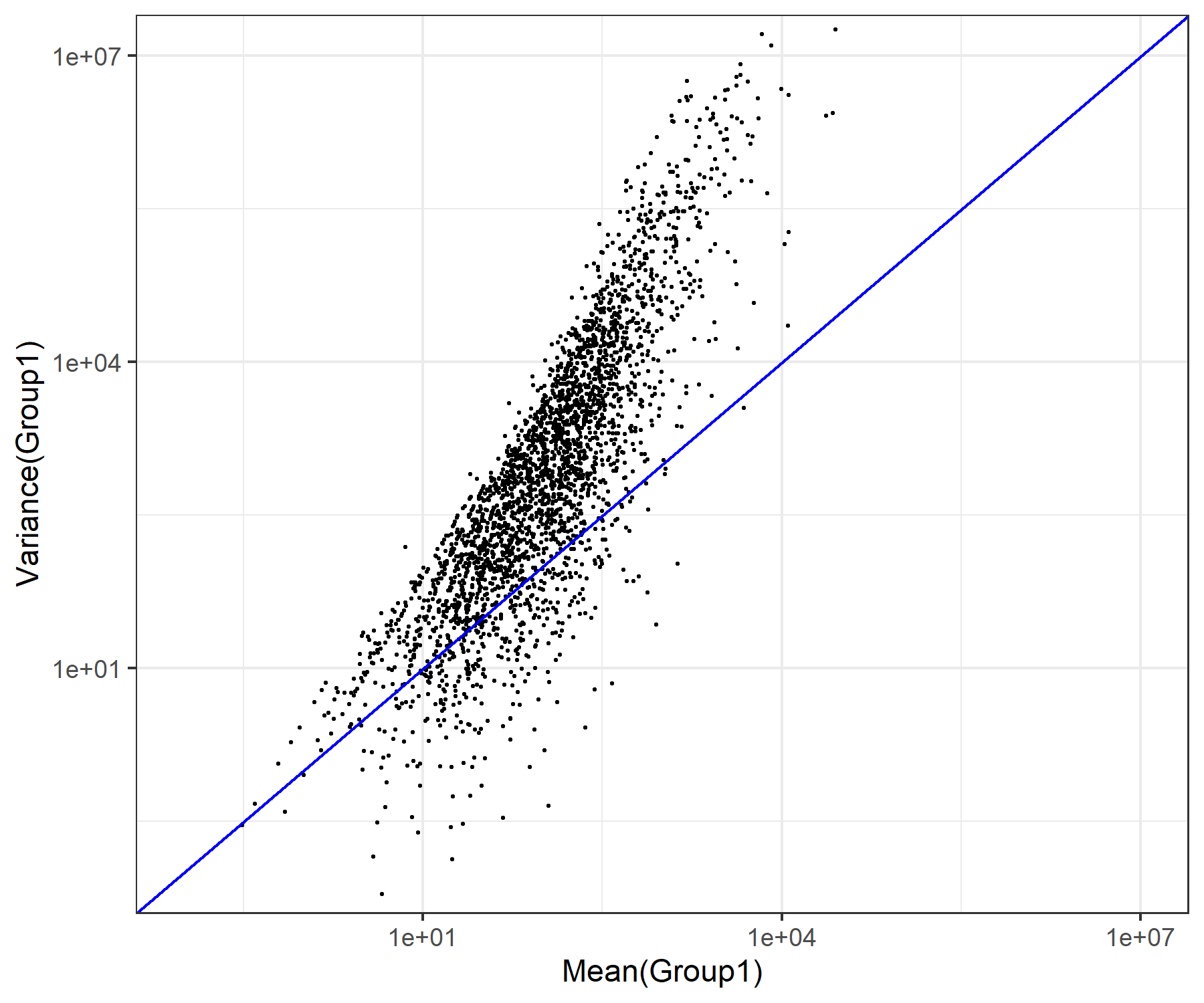{kind=link}
W8.10 回帰モデル(回帰直線や回帰曲線)
近似曲線や近似直線を引くやり方です。geom_smoothやstat_smoothが利用可能です。
W8.10.1 geom_smoothを利用
geom_smoothを利用するやり方です。回帰直線にしたい場合はmethod="lm"とやればよいです。デフォルトは95%信頼区間を灰色で示してくれるのがse=TRUEで、信頼区間を表示させたくない場合はse=FALSEとやります。この場合は95%信頼区間の幅が非常に狭くわかりづらいのですが、一応表示されています。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)") +
geom_smooth(method="lm", se=TRUE, color="black")
plot(g)
W8.10.2 stat_smoothを利用
stat_smoothを利用するやり方です。基本的にgeom_smoothと同じ結果が得られるので、color="red"にしたり、99.99%信頼区間として灰色の領域を広く示そうとしています。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)") +
stat_smooth(method="lm", se=TRUE, color="red", level=0.9999)
plot(g)
W8.10.3 デフォルトのloess曲線
stat_smoothを利用しつつ、methodオプションを指定せずに、デフォルトのloess曲線を利用するやり方です。これだと、右上や左下の灰色で示されている信頼区間がデフォルトの95%でも広がっていてわかりやすいですね。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)") +
stat_smooth()
plot(g)
W8.10.4 一般化線形モデル
stat_smoothを利用しつつ、method="glm"として一般化線形モデル(Generalized
Linear Moldel; GLM)を利用するやり方です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN1,y=VAR1)) +
geom_point(shape=20, size=0.6, color="black") +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean(Group1)",y="Variance(Group1)") +
stat_smooth(method="glm", family="Gamma", color="black", level=0.99)
plot(g)
描画:ggplot2の本番(W9)
ggplot2を用いて描画するやり方を示します。さきほどのW8では、全部で3群(G1群、G2群、G3群)あるデータのうち、G1群(酸ストレス短期暴露群;pH4.5_1h群)に限定し、基本的な使い方をメインに解説しました。ここでは、3群全てのデータを入力として用い、本文中の図4の作成を詳細に解説します。R再起動後でもリカバリできるように、入力ファイルの読込から群ごとのカウントの平均と分散の作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
M <- NULL
V <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
}W9.1 入力データの準備
ggplot2は、入力としてデータフレーム形式のオブジェクトを用いるのが一般的であり、列名を指定してプロットに用いる情報を指定します。W8.1と同様、平均Mと分散Vの情報をベースとするのは基本ですが、3群全ての情報を含むため、「平均の列(MEAN)、分散の列(VARIANCE)、そしてどの行がどの群に属するかを表す群ラベル情報の列(GROUP)」という計3列に収めてデータを格納します。
W9.1.1 行列をベクトルに変換
上記の計3列にするためには、まず2949行×3列のMとVを2949×3
=
8847個の要素(元)からなるベクトルにそれぞれ変換しておく必要があります。これを行うための関数がas.vectorです。これは文字通り「ベクトルとして(as
vector)」という意味です。ここでは、as.vector(M)の結果が行列ではない(つまりNULL)であることをまず示し、次に、W3.2でも紹介したベクトルの要素数をカウントするlength関数を実行しています。最後にhead(M)とhead(as.vector(M))を表示させて概要を把握しています。ちなみにVも同じなので割愛します。
dim(M)## [1] 2949 3dim(as.vector(M))## NULLlength(as.vector(M))## [1] 8847head(M)## [,1] [,2] [,3]
## EBG00001128470 219.35372 139.9754 163.20042
## EBG00001128476 88.02994 116.9911 45.61295
## EBG00001128500 26661.18736 23549.0411 35571.20466
## EBG00001128509 259.62436 166.7145 190.11327
## EBG00001128529 0.00000 0.0000 0.00000
## LGG_00001 419.03678 412.6478 318.10139head(as.vector(M))## [1] 219.35372 88.02994 26661.18736 259.62436 0.00000 419.03678W9.1.2 クラスラベル作成の基本形
W5.1と似たノリお部分ですが、ここでやりたいのは、行列データのどの列(サンプル)がどの群に属するかを指し示すものではなく、ベクトルデータのどの要素(遺伝子)がどの群に属するかを指定するのが目的です。もちろん最終的には行列データにするのですが、その場合でもどの行(遺伝子)がどの群に属するかを指定するのに相当します。ここでは、mapply関数を用いて群数Kまでの整数ベクトル(つまり1:K)を、それぞれの要素(つまり1,
2,
3)につきnrow(M)個だけ生成させ、最終的にそれらをベクトルに型変換して(as.vector)、groupというオブジェクトに格納しています。ちょっとややこしいのですが、要するに「1が2949個、2が2949個、3が2949個」という計8847個の要素からなる整数ベクトルがgroupの実体だということです。念のためgroupの要素数をlength関数で調べるとともに、table関数でどの要素が何個存在するかを表示させています。table関数については、念のため挙動がわかる他のベクトルの実行例も示しています。
K## [1] 3group <- as.vector(mapply(rep, 1:K, nrow(M)))
length(group)## [1] 8847head(group)## [1] 1 1 1 1 1 1table(group)## group
## 1 2 3
## 2949 2949 2949table(c("C", "uge", "C", 5, "C", "C", 2, 4, 5))##
## 2 4 5 C uge
## 1 1 2 4 1W9.1.3 クラスラベル作成の本番
W9.1.2でもよいのですが、ggplot2で作図する場合は、このgroupで作成したものがそのまま凡例として表示されます。それゆえ、この段階で作図を意識して文字列にしておきます。具体的にはmapplyのところで1:Kとしていた部分を”G1(pH4.5_1h)“,”G2(pH4.5_24h)“,”G3(pH7_CCG)“という文字列にしておきます。
K## [1] 3hoge <- c("G1(pH4.5_1h)", "G2(pH4.5_24h)", "G3(pH7_CCG)")
group <- as.vector(mapply(rep, hoge, nrow(M)))
length(group)## [1] 8847head(group)## [1] "G1(pH4.5_1h)" "G1(pH4.5_1h)" "G1(pH4.5_1h)" "G1(pH4.5_1h)" "G1(pH4.5_1h)"
## [6] "G1(pH4.5_1h)"table(group)## group
## G1(pH4.5_1h) G2(pH4.5_24h) G3(pH7_CCG)
## 2949 2949 2949W9.1.4 入力データ作成本番
いよいよ、W9.1のところで述べた、「平均の列(MEAN)、分散の列(VARIANCE)、そしてどの行がどの群に属するかを表す群ラベル情報の列(GROUP)」という計3列に収めたデータフレーム形式のオブジェクトdを作成します。data.frame関数実行部分は、見やすいように3行に分けています。「MEAN,
VARIANCE,
GROUP」という大文字部分が、最終的に得られるデータフレーム形式の行列dの列名として与えているものになります。確かに8847行×3列になっていて、3列目を見たときに、最初の6行分を表示するheadでは”G1(pH4.5_1h)“、最後の6行分を表示するtailでは”G3(pH7_CCG)“になっていて妥当ですね。
d <- data.frame(MEAN = as.vector(M),
VARIANCE = as.vector(V),
GROUP = group)
dim(d)## [1] 8847 3head(d)## MEAN VARIANCE GROUP
## 1 219.35372 13304.9755 G1(pH4.5_1h)
## 2 88.02994 2165.1651 G1(pH4.5_1h)
## 3 26661.18736 2736641.8214 G1(pH4.5_1h)
## 4 259.62436 29046.4243 G1(pH4.5_1h)
## 5 0.00000 0.0000 G1(pH4.5_1h)
## 6 419.03678 906.3969 G1(pH4.5_1h)tail(d)## MEAN VARIANCE GROUP
## 8842 11.57086 1.692079e+01 G3(pH7_CCG)
## 8843 389.06316 3.615898e+03 G3(pH7_CCG)
## 8844 278.79022 3.723977e+02 G3(pH7_CCG)
## 8845 89.72799 1.443510e+02 G3(pH7_CCG)
## 8846 963.61110 4.679214e+04 G3(pH7_CCG)
## 8847 11687.93670 3.189402e+07 G3(pH7_CCG)W9.2 群ごとに分ける
W9.2.1 プロットの色で分ける
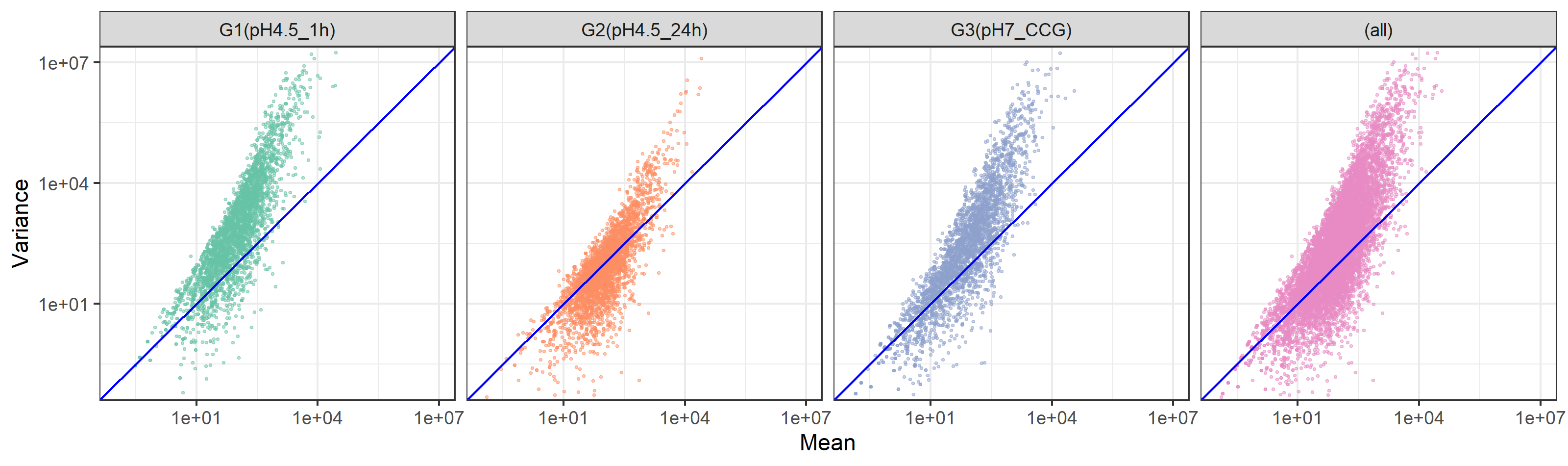
群ごとに色分けするために必要な情報は、dオブジェクトのGROUP列に格納されています。以下のスクリプトは、W8.9.3をベースとしていますが、dオブジェクトの列名が違いますので、aesの中身をx=MEAN, y=VARIANCE, color=GROUPに変更しています。また、geom_pointにつけていたオプションは消し、labsの内容もGroup1に限定したものではないので変更しています。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance")
plot(g)
W9.2.2 プロットの形で分ける
プロットの形で分ける基本形は、aesオプションのところでcolor=GROUPだったところをshape=GROUPにするだけです。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, shape=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance")
plot(g)
W9.2.3 プロットを群ごとに色で分ける(行方向)
群ごとに独立してプロットするやり方です。3行1列っぽくしたい場合は、facet_gridを使って、rowsオプションをつけるのが基本形です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(rows=vars(GROUP))
plot(g)
W9.2.4 プロットを群ごとに色で分ける(列方向)
群ごとに独立してプロットするやり方です。1行3列っぽくしたい場合は、facet_gridを使って、colsオプションをつけるのが基本形です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP))
plot(g)
W9.2.5 プロットを群ごとに色で分ける(2行2列)
群ごとに独立してプロットするやり方です。2行2列っぽくしたい場合は、facet_wrapを使います。ここで指定しているオプションのvars(GROUP)は、入力オブジェクト中のGROUP列で分けよという指定であり、~GROUPと指定してもかまいません。ncol=2というのは、列数を2にせよという意味です。今は群数が3しかないことがわかっていますので、最初の群(つまりG1)が1行1列目の箇所に、2番目の群(つまりG2)が1行2列目の箇所にプロットされた段階で、3番目の群(つまりG3)は次の行(つまり2行1列目)になります。もし群数が5だと、3行2列のプロットになります。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_wrap(vars(GROUP), ncol=2)
plot(g)
W9.2.6 プロットを群ごとに色で分ける(1行3列)
さきほどのW9.2.5の説明から予想できますが、facet_wrap(~GROUP, ncol=3)とやると、W9.2.4のfacet_grid(cols=vars(GROUP))と同じ結果になります。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_wrap(vars(GROUP), ncol=3)
plot(g)
W9.2.7 群ごとだけでなく全てをマージしたものも追加する(1行4列)
W9.2.4において、facet_gridのところでmargins=TRUEを追加すると、全ての群をマージしたものも追加でプロットできます。(本当はfacet_wrapを用いて2行2列で収めたかったのですが、どうやらこの関数ではできなそうです…)。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
plot(g)
W9.3 凡例を消す
W9.3.1 やり方1(geom_point内のオプションを利用)
上図はわざわざ凡例(legend)を表示させなくてもよい場合だと思います。このような場合に、geom_pointの中で、show.legend = FALSEを与えることで目的を達成するやり方です。こうすることで、少し横幅が広がるメリットがあります。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
plot(g)
W9.3.2 やり方2(themeを利用)
geom_pointはいじらずに、theme(legend.position="none")を追加するやり方です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point() +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE) +
theme(legend.position="none")
plot(g)
W9.4 半透明のプロットにする
geom_pointで数万個程度の点からなる散布図になると、点がある程度大きい場合に重なりまくって密度がよくわからないという問題が発生します。これを回避する手段として、点の透過率を調整するやり方があります。スクリプトはW9.3.1をベースにしています。
W9.4.1 透過率40%(alpha=0.4)の場合
点を半透明にしたい場合は、geom_point内のalphaというオプションで与えます。たとえば40%の透過率にしたい場合はalpha=0.6を指定します。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.6) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
plot(g)
W9.4.2 透過率70%(alpha=0.3)の場合
70%の透過率にしたい場合はalpha=0.3を指定します。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.3) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
plot(g)
W9.4.3 透過率90%(alpha=0.1)の場合
90%の透過率にしたい場合はalpha=0.1を指定します。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.1) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
plot(g)
W9.4.4 透過率70%で、点の形や大きさも変更する
これはW8.7との組み合わせになります。思考回路としては、「点のサイズを小さくするのを併用すれば、さらに重なりを回避できる」です。たとえば、shape=19だったのをshape=20にするだけで、点の大きさが半分ほどになります。これはシンプルに両者の点の形状が同じで大きさが異なるだけだからです。ここではgeom_point内でshape=19およびsize=0.7の場合の結果を示します。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.3, shape=19, size=0.7) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
plot(g)
W9.4.5 透過率55%で、点の形や大きさも変更する
これはW8.7との組み合わせになります。基本的な思考回路としては、「点のサイズを小さくするのを併用すれば、さらに重なりを回避できる」です。たとえば、shape=19だったのをshape=20にするだけで、点の大きさが半分ほどになります。これはシンプルに両者の点の形状が同じで大きさが異なるだけだからです。ここではgeom_point内でshape=19およびsize=0.4の場合の結果を示します。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
plot(g)
W9.5 図のサイズを変更
基本的にW8.9.3との組み合わせになります。現状各プロットが縦長になっているので、全体として横長にすることで、各プロットを正方形に近づけようとしていますす。これはy=xの直線が斜め45度にしたいという思想です。W9.4.5をベースとしています。
W9.5.1 2200×800ピクセルのpngファイル
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
ggsave("JSLAB20.w9.5.1.png", g, units="px", width=2200, height=800, device="png")得られたJSLAB20.w9.5.1.pngは、以下に見えているものと同じになります。

{kind=link}
W9.5.2 2630×800ピクセルのpngファイル
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
ggsave("JSLAB20.w9.5.2.png", g, units="px", width=2630, height=800, device="png")得られたJSLAB20.w9.5.2.pngは、以下に見えているものと同じになります。

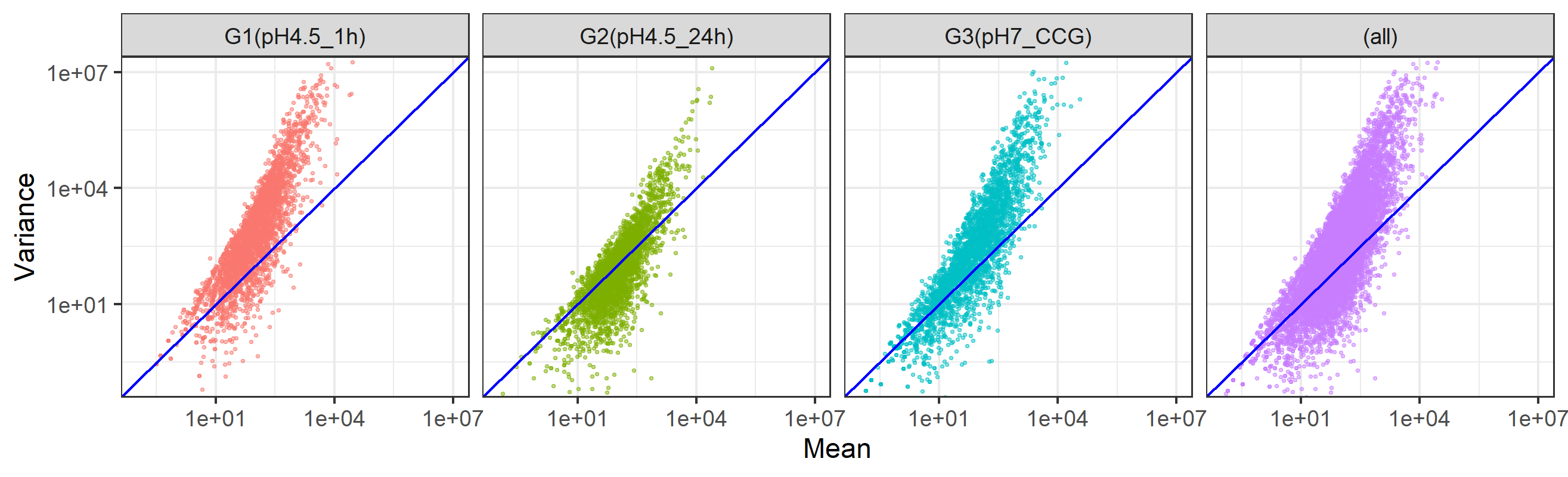{kind=link}
W9.5.3 3200×950ピクセルのpngファイル
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
ggsave("JSLAB20.w9.5.3.png", g, units="px", width=3200, height=950, device="png")得られたJSLAB20.w9.5.3.pngは、以下に見えているものと同じになります。

{kind=link}
W9.6 プロットの色を変更
W9.5.3までの結果だと、群ごとに任意の色を指定できないので、そのあたりを自在に行うやり方を示します。たとえばhttp://www.stat.columbia.edu/~tzheng/files/Rcolor.pdfなどで色の見本が提供されています。R再起動後でもリカバリできるように、入力ファイルの読込から群ごとのカウントの平均と分散の作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
M <- NULL
V <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
}
hoge <- c("G1(pH4.5_1h)", "G2(pH4.5_24h)", "G3(pH7_CCG)")
group <- as.vector(mapply(rep, hoge, nrow(M)))
d <- data.frame(MEAN = as.vector(M),
VARIANCE = as.vector(V),
GROUP = group)W9.6.1 点を全て黒にする
W9.5.3をベースとして、まずはaesのところのcolor=GROUPを消して、点を全て黒にするやり方です。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE)
ggsave("JSLAB20.w9.6.1.png", g, units="px", width=3200, height=950, device="png")得られたJSLAB20.w9.6.1.pngは、以下に見えているものと同じになります。

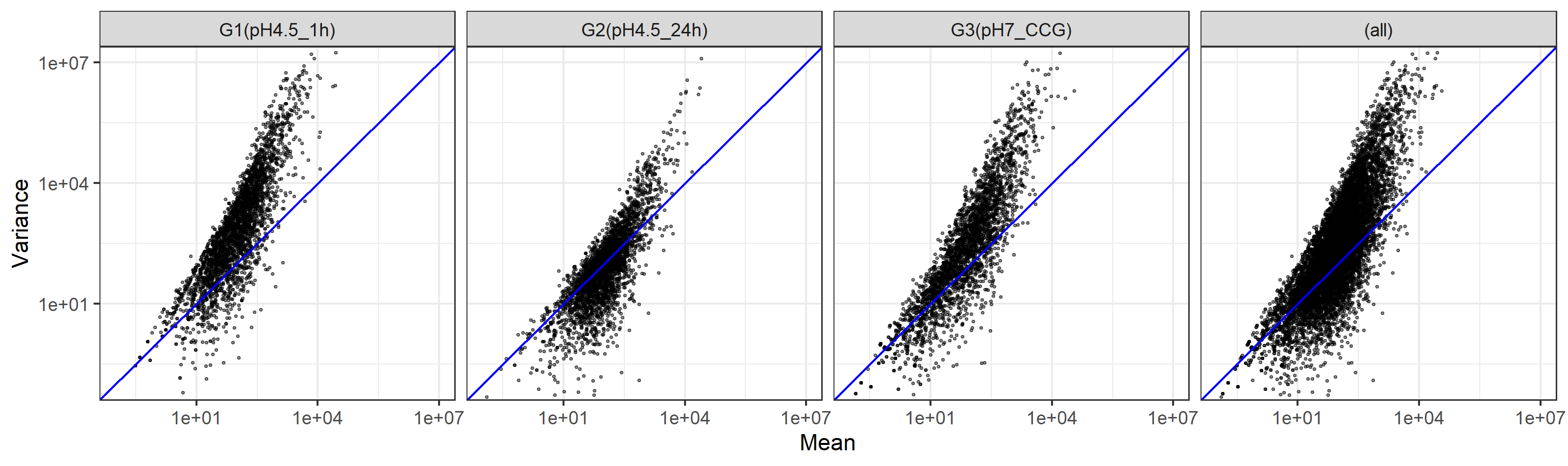{kind=link}
W9.6.2 RColorBrewerカラーパレットを利用
RColorBrewerが提供するカラーパレットを利用するやりかたです。まずはどのようなカラーパレットを指定可能かを示します。左側に見えているものがパレット名です。
RColorBrewer::display.brewer.all()
ここではW9.5.3をベースとして、scale_color_brewer(palette="Set2")を実行しています。他には、たとえば実験デザインが「薬剤A未処理群と処理群、薬剤B未処理群と処理群」のようなペアっぽくなっている場合は、scale_color_brewer(palette="Paired")とかにしてもよいと思います。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE) +
scale_color_brewer(palette="Set2")
ggsave("JSLAB20.w9.6.2.png", g, units="px", width=3200, height=950, device="png")得られたJSLAB20.w9.6.2.pngは、以下に見えているものと同じになります。

{kind=link}
W9.6.3 マニュアルで色指定
マニュアルで色を指定したい場合はscale_color_manualを利用します。ここでは群1が”coral”、群2が”olivedrab3”、群3が”red”、そして全てをマージしたものが”black”となるように指定しています。色見本はhttp://www.stat.columbia.edu/~tzheng/files/Rcolor.pdfなどで提供されています。
axis_range <- c(1e-01, 1e+7)
g <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
facet_grid(cols=vars(GROUP), margins=TRUE) +
scale_color_manual(values=c("coral", "olivedrab3", "red", "black"))
ggsave("JSLAB20.w9.6.3.png", g, units="px", width=3200, height=950, device="png")得られたJSLAB20.w9.6.3.pngは、以下に見えているものと同じになります。

{kind=link}
W9.7 複数プロットのレイアウト
実用上はW9.6まで頑張り、あとはパワポなどで手作業でレイアウトするのでも十分です。しかし、もう少し頑張れば、たとえばW9.2.5で示したような2行2列で、最後の右下部分に全データの図を配置したりできます。ここでは、複数の図をうまくレイアウトするpatchworkパッケージを利用するやり方を示します。R再起動後でもリカバリできるように、入力ファイルの読込から群ごとのカウントの平均と分散の作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
M <- NULL
V <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
}
hoge <- c("G1(pH4.5_1h)", "G2(pH4.5_24h)", "G3(pH7_CCG)")
group <- as.vector(mapply(rep, hoge, nrow(M)))
d <- data.frame(MEAN = as.vector(M),
VARIANCE = as.vector(V),
GROUP = group)W9.7.1 patchworkのインストールとロード
install.packages("patchwork")
library(patchwork)W9.7.2 レイアウトしたい個々の描画用オブジェクトの作成
ここでは、G1のみ、G2のみ、G3のみ、そして全データの計4つの描画用オブジェクトを作成します。W9.6.3をベースとしていますが、スクリプト下部にあったfacet_gridやscale_color_manualは削除しています。まずはG1群からです。冒頭部分でgrep関数を用いて、行列d中の”GROUP”列を入力として、"G1"という文字列を含むインデックス情報(行番号)を取得した結果をobjに格納しています。次に、行列dの中からobjで指定した行のみを抽出した結果をd1に格納しています。ggplotはこのd1を入力として行い、結果をg1に格納しています。これでG1群のみのプロットに必要な情報がg1に格納されたことになります。
coloor <- "magenta"
obj <- grep("G1", d[, "GROUP"])
d1 <- d[obj, ]
axis_range <- c(1e-01, 1e+7)
g1 <- ggplot(d1, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4, col=coloor) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance")
plot(g1)
G2についても同様に色だけ変更してg2オブジェクトに格納しておきます。
coloor <- "navy"
obj <- grep("G2", d[, "GROUP"])
d2 <- d[obj, ]
axis_range <- c(1e-01, 1e+7)
g2 <- ggplot(d2, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4, col=coloor) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance")
plot(g2)
G3についても同様に色だけ変更してg3オブジェクトに格納しておきます。
coloor <- "orange"
obj <- grep("G3", d[, "GROUP"])
d3 <- d[obj, ]
axis_range <- c(1e-01, 1e+7)
g3 <- ggplot(d3, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4, col=coloor) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance")
plot(g3)
全データについてはサブセットにする必要はないのでそのままdオブジェクトを入力として、色だけ変更してg_allオブジェクトに格納しておきます。
coloor <- "gray31"
axis_range <- c(1e-01, 1e+7)
g_all <- ggplot(d, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4, col=coloor) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance")
plot(g_all)
W9.7.3 レイアウト本番
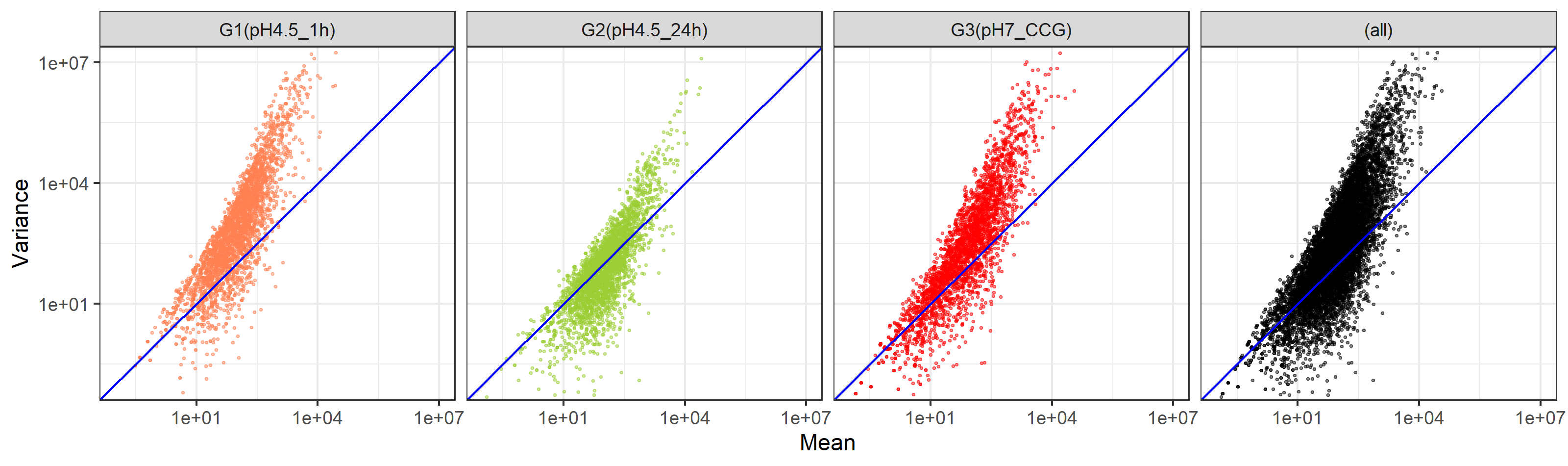
作成済みの描画用オブジェクトであるg1、g2、g3、g_allが4つ存在するという前提で行います。基本的には表示させたい順に記載していくだけです。最後にplot_layout(ncol=2)とやっていますが、これは列数を2つに限定せよという指令です。
library(patchwork)
g1 + g2 + g3 + g_all + plot_layout(ncol=2)
以下はplot_layout(ncol=1)として列数を1つに限定しつつ、g3,
g2の順番で2つだけプロットをせよという指令です。
g3 + g2 + plot_layout(ncol=1)
以下はplot_layout(ncol=2)として列数を2つに限定しつつ、g3,
g1, g1, g2,
g_allの順番でプロットせよという指令です。こんな感じで、同じものを重複させることもできます。
g3 + g1 + g1 + g2 + g_all + plot_layout(ncol=2)
W9.8 文字列の追加
ここでは、W9.7のプロット上にテキストを追加するやり方を示します。これは注釈付け(アノテーション)ともいうことを踏まえると、実際に用いる関数名がannotateである理由が納得しやすいと思います。実際に行うのは、W7.6でも調べた「平均Mのほうが分散Vよりも大きい遺伝子数を右下、その逆を左上に表示」です。R再起動後でもリカバリできるように、入力ファイルの読込から群ごとのカウントの平均と分散の作成までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
M <- NULL
V <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
}
hoge <- c("G1(pH4.5_1h)", "G2(pH4.5_24h)", "G3(pH7_CCG)")
group <- as.vector(mapply(rep, hoge, nrow(M)))
d <- data.frame(MEAN = as.vector(M),
VARIANCE = as.vector(V),
GROUP = group)
obj <- grep("G1", d[, "GROUP"])
d1 <- d[obj, ]
obj <- grep("G2", d[, "GROUP"])
d2 <- d[obj, ]
obj <- grep("G3", d[, "GROUP"])
d3 <- d[obj, ]W9.8.1 レイアウトしたい個々の描画用オブジェクトの作成
W9.7.1と同じノリで、まずはG1から作成し、g1オブジェクトに格納しておきます。
coloor <- "magenta"
hoge <- d1
Vlow <- sum(hoge[,1] > hoge[,2])
Vhigh <- sum(hoge[,1] < hoge[,2])
axis_range <- c(1e-01, 1e+7)
g1 <- ggplot(d1, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4, col=coloor) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
annotate("text", x=1e+06, y=1e+00, label=Vlow) +
annotate("text", x=1e+00, y=1e+06, label=Vhigh)
plot(g1)
G2についてもg2オブジェクトに格納しておきます。
coloor <- "navy"
hoge <- d2
Vlow <- sum(hoge[,1] > hoge[,2])
Vhigh <- sum(hoge[,1] < hoge[,2])
axis_range <- c(1e-01, 1e+7)
g2 <- ggplot(d2, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4, col=coloor) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
annotate("text", x=1e+06, y=1e+00, label=Vlow) +
annotate("text", x=1e+00, y=1e+06, label=Vhigh)
plot(g2)
G3についてもg3オブジェクトに格納しておきます。
coloor <- "orange"
hoge <- d3
Vlow <- sum(hoge[,1] > hoge[,2])
Vhigh <- sum(hoge[,1] < hoge[,2])
axis_range <- c(1e-01, 1e+7)
g3 <- ggplot(d3, aes(x=MEAN, y=VARIANCE)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4, col=coloor) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
annotate("text", x=1e+06, y=1e+00, label=Vlow) +
annotate("text", x=1e+00, y=1e+06, label=Vhigh)
plot(g3)
全データについてもg_allオブジェクトに格納しておきます。色指定についてはW9.7.2とはちょっと変えて、W9.6.3で使ったscale_color_manualを流用して、各群由来の色を重ね書きするような感じにしています。
hoge <- d
Vlow <- sum(hoge[,1] > hoge[,2])
Vhigh <- sum(hoge[,1] < hoge[,2])
axis_range <- c(1e-01, 1e+7)
g_all <- ggplot(d, aes(x=MEAN, y=VARIANCE, color=GROUP)) +
geom_point(show.legend = FALSE, alpha=0.45, shape=19, size=0.4) +
scale_x_log10() +
scale_y_log10() +
theme_bw() +
geom_abline(intercept=0, slope=1, color="blue") +
coord_cartesian(xlim=axis_range, ylim=axis_range) +
labs(x="Mean",y="Variance") +
annotate("text", x=1e+06, y=1e+00, label=Vlow) +
annotate("text", x=1e+00, y=1e+06, label=Vhigh) +
scale_color_manual(values=c("magenta", "navy", "orange"))
plot(g_all)
W9.8.2 レイアウト本番1
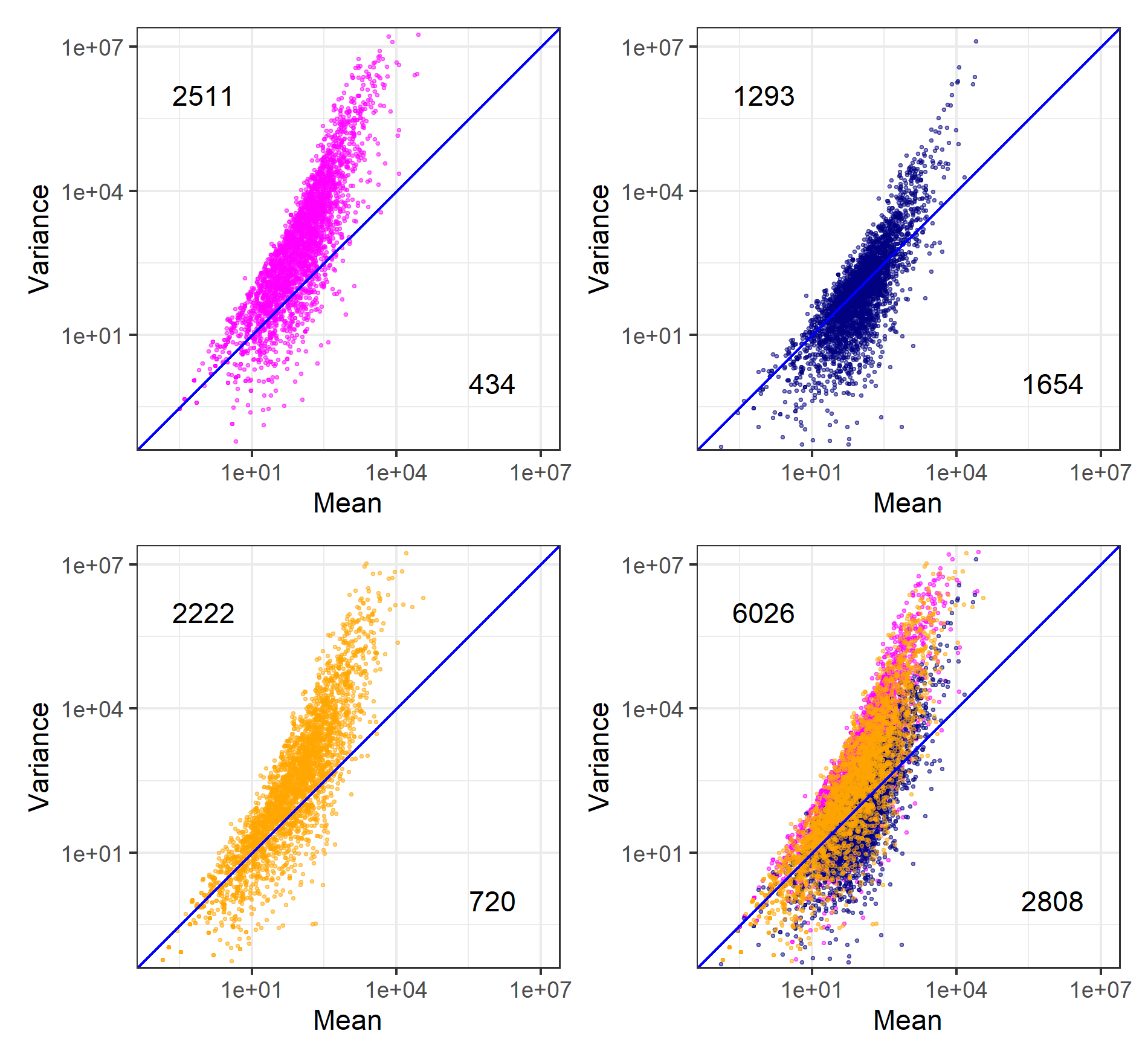
作成済みの描画用オブジェクトであるg1、g2、g3、g_allが4つ存在するという前提で行います。基本的には表示させたい順に記載していくだけです。最後にplot_layout(ncol=2)とやっていますが、これは列数を2つに限定せよという指令です。この結果をみてもわかりますが、各プロットが小さくなる一方で、プロットに追加した右下と左上の数値の大きさは不変です。したがって、このあたりは結果を見ながらアドホックにannotate関数のxとyの再指定(ちなみにこの座標はテキスト中心の位置の指定です)を行う感じになります。
g1 + g2 + g3 + g_all + plot_layout(ncol=2)
W9.8.3 レイアウト本番2
今度はpngファイルで保存するやり方です。W9.6でも使ったggsave関数を利用しています。これが図4の原形です。
g <- g1 + g2 + g3 + g_all + plot_layout(ncol=2)
ggsave("JSLAB20.w9.8.3.png", g, units="px", width=1900, height=1760, device="png")得られたJSLAB20.w9.8.3.pngは、以下に見えているものと同じになります。

{kind=link}
W10 様々な確認
R再起動後でもリカバリできるように、入力ファイルの読込から群ごとのカウントの平均・分散・dispersionの計算までの必要最小限のスクリプトを再掲しておきます。ここは改めて実行しても時間的には誤差範囲ですので、実行は任意です。
library(openxlsx)
A <- read.xlsx("JSLAB18.xlsx", rowNames=TRUE)
T <- colSums(A)
Anorm <- sweep(x=A, MARGIN=2, STATS=1000000/T, FUN="*")
nk <- c(3, 3, 3)
K <- length(nk)
L <- as.vector(mapply(rep, 1:K, nk))
func_phi <- function(x){
if(mean(x) == 0){ phi_value <- 0 }
else{ phi_value <- (var(x) - mean(x))/mean(x)^2 }
return(phi_value)
}
M <- NULL
V <- NULL
Phi <- NULL
for(i in 1:K){
M <- cbind(M, apply(X=Anorm[, L==i], MARGIN=1, FUN=mean))
V <- cbind(V, apply(X=Anorm[, L==i], MARGIN=1, FUN=var))
Phi <- cbind(Phi, apply(X=Anorm[, L==i], MARGIN=1, FUN=func_phi))
}W10.1 dispersionパラメータの要約統計量
群ごとの要約統計量です。特にG2については、\(\phi_{i,2}\)の平均値がマイナス(\(= -0.002631\))になっていることがわかります。なお、\(\phi_{i,2}\)の中央値もマイナス(\(= -0.000909\))ですが、これは図4の原形であるW9.8.3で見えている\(y = x\)の左上側(=1293個)と右下側(= 1654個)の遺伝子数分布から明らかです。
summary(Phi)## V1 V2 V3
## Min. :-0.63080 Min. :-4.923486 Min. :-4.145943
## 1st Qu.: 0.01220 1st Qu.:-0.007622 1st Qu.: 0.000234
## Median : 0.07186 Median :-0.000909 Median : 0.029073
## Mean : 0.16608 Mean :-0.002631 Mean : 0.085029
## 3rd Qu.: 0.22203 3rd Qu.: 0.005891 3rd Qu.: 0.114288
## Max. : 2.86008 Max. : 1.679419 Max. : 2.725156W10.2 サンプル間クラスタリング
乳酸菌NGS連載第18回の図7と同じものです。このクラスタリング結果からも、群2(pH4.5_24h)の反復データ間の類似度が非常に高い(距離が短い)ことがわかります。
data <- A
out <- clusterSample(data, dist.method="spearman",
hclust.method="average", unique.pattern=TRUE)
plot(out)